An Amperometric Biosensor for the Determination of Bacterial Sepsis Biomarker, Secretory Phospholipase Group 2-IIA Using a Tri-Enzyme System



Abstract
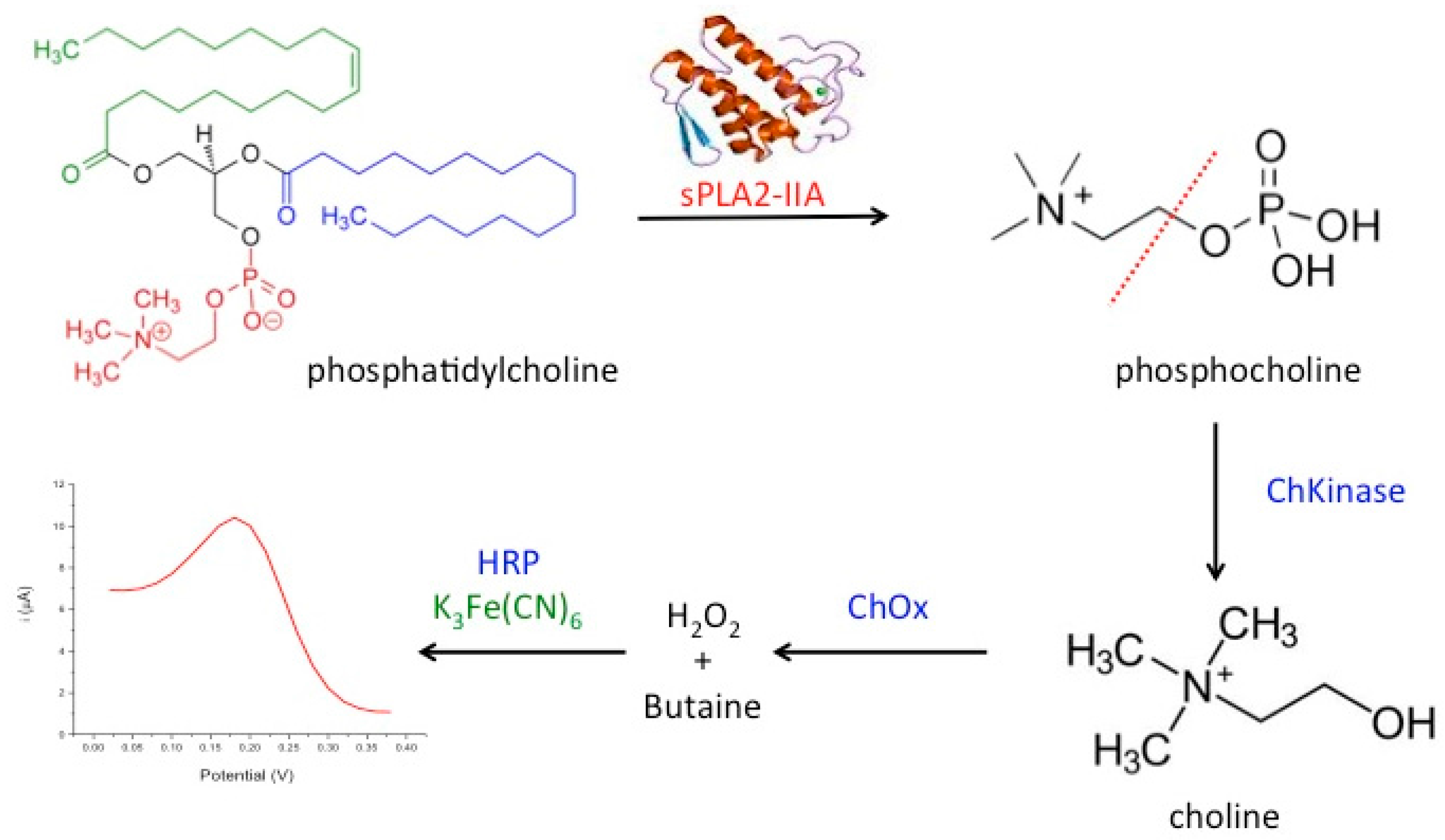
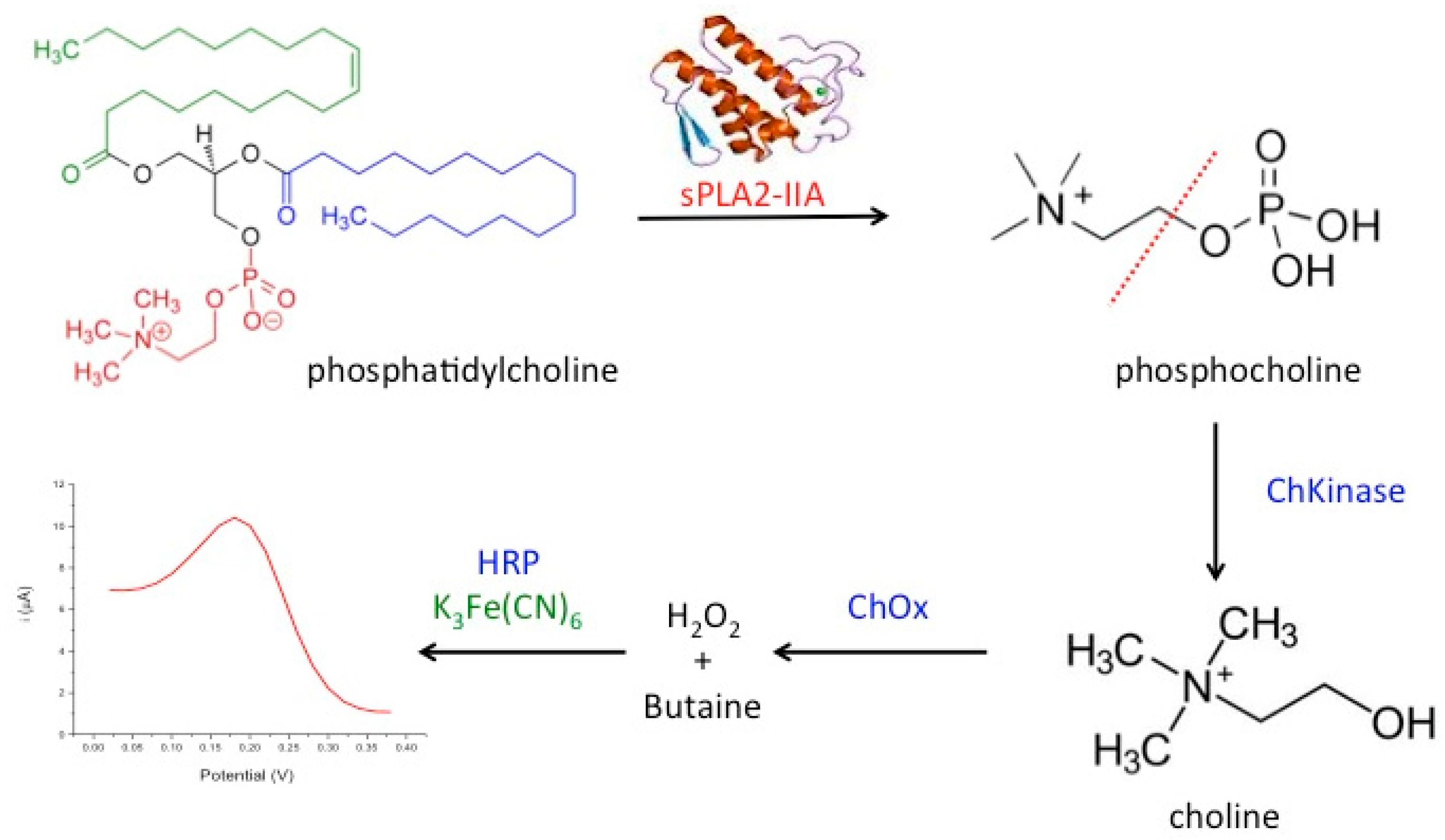
:1. Introduction
2. Materials and Methods
2.1. Reagents and Solutions
2.2. Apparatus and Electrode Deionized Water Was Used
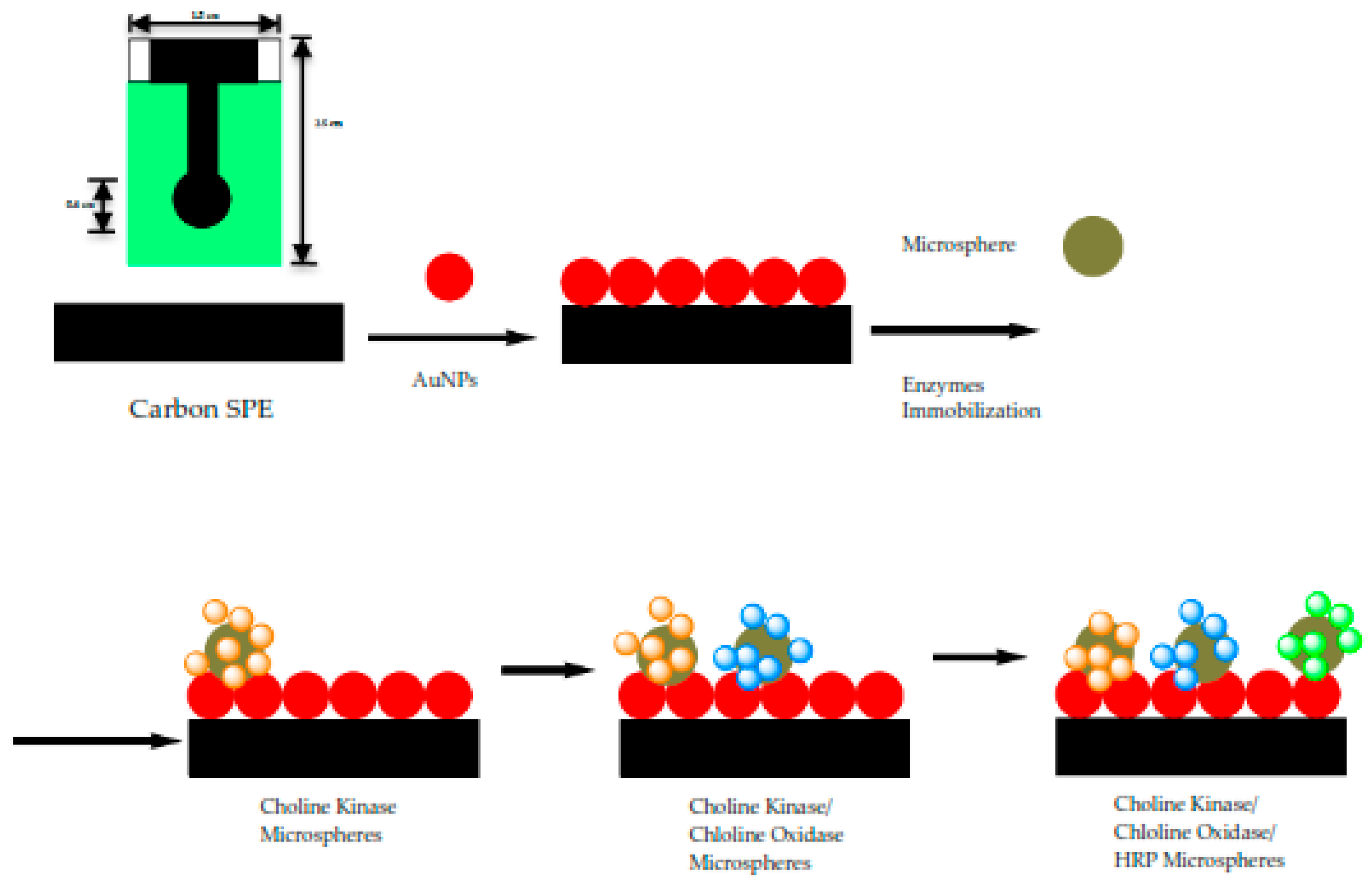
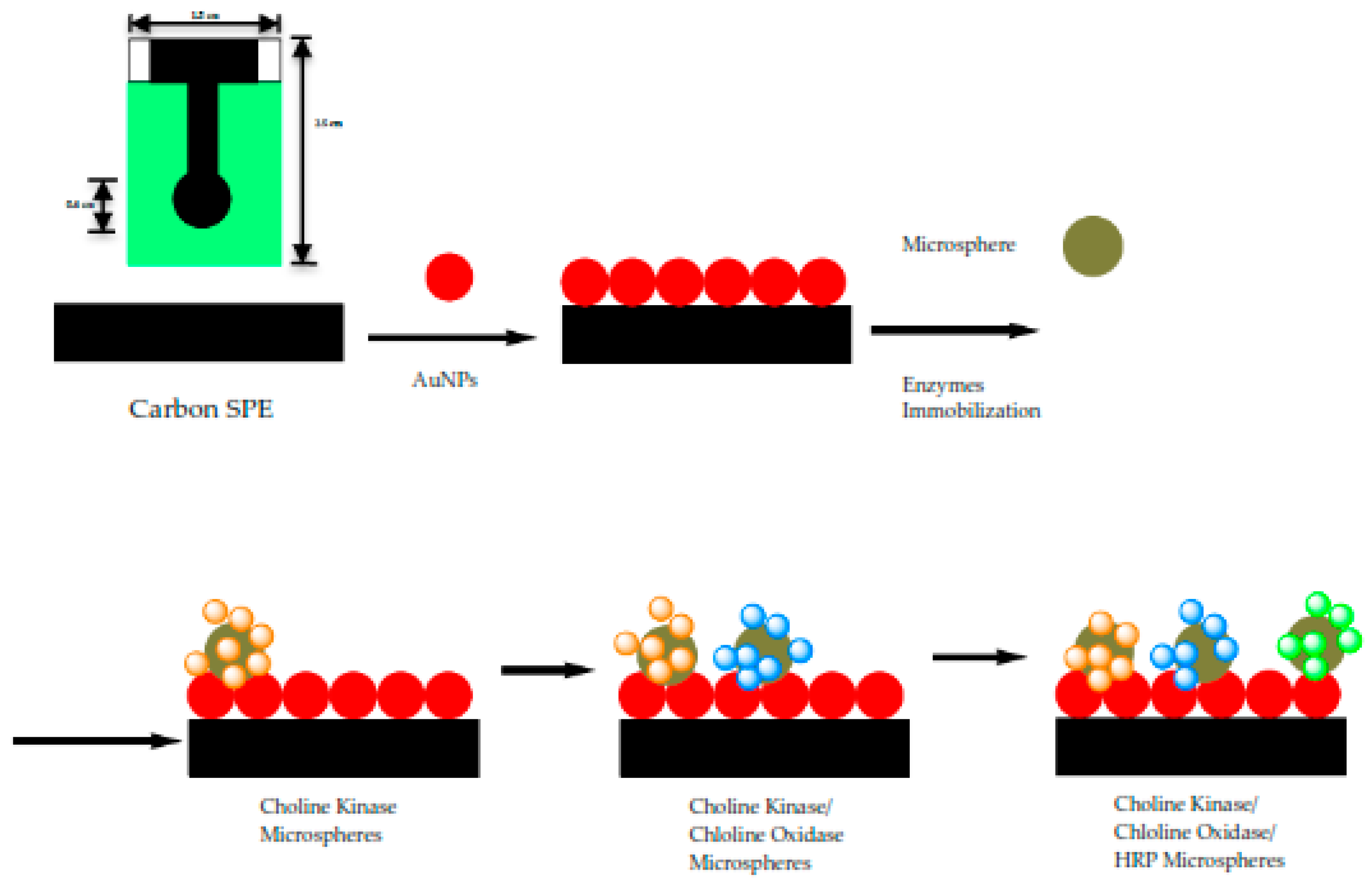
2.3. Immobilization Matrix—Acrylic Microspheres
2.4. Preparation of Modified SPEs
2.5. Optimization of Bacterial Sepsis Biosensor
2.6. Selectivity Response
2.7. Amperometric Measurement
2.8. Human Blood Serum Analysis
3. Results and Discussion
3.1. FESEM Measurement of ACMS and Modified SPEs
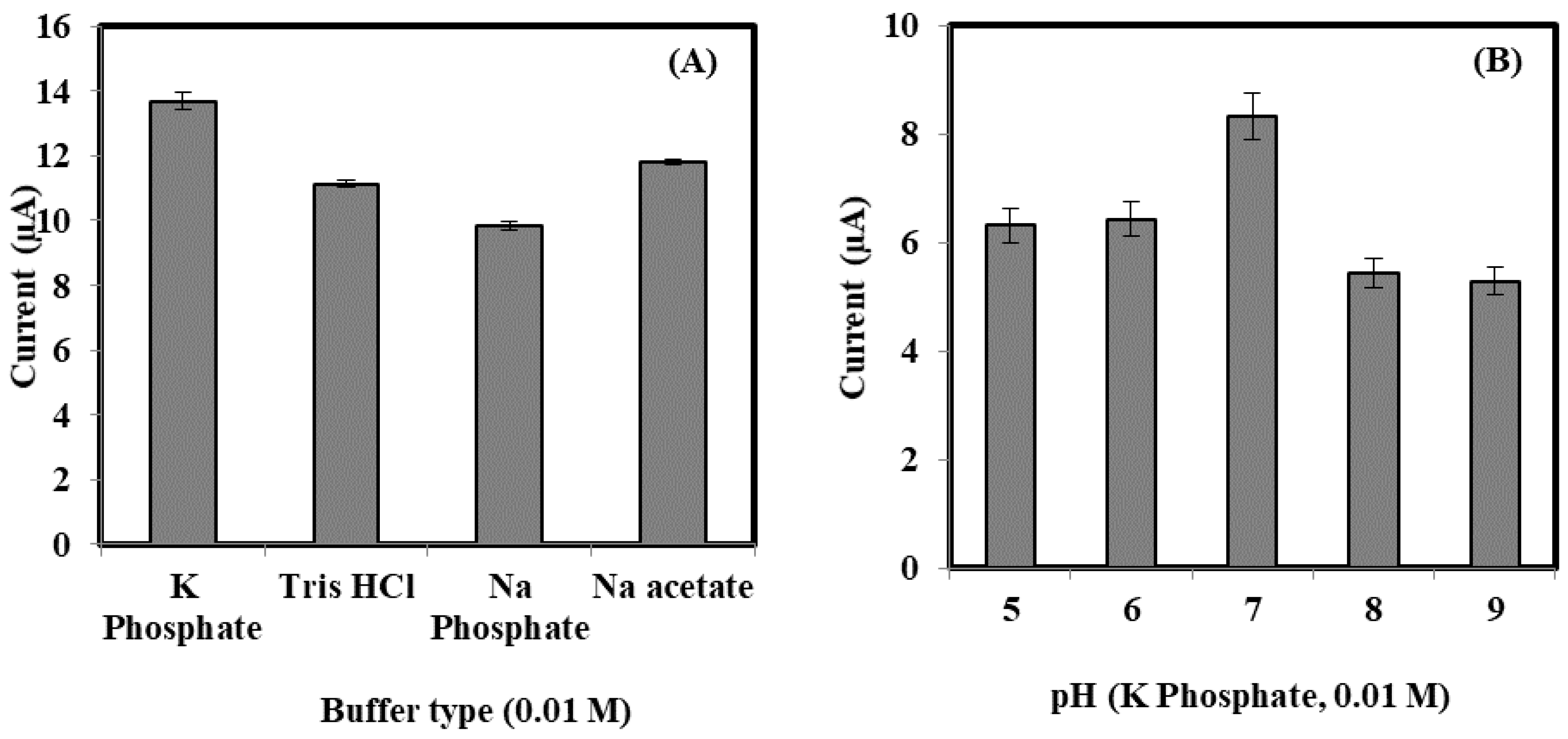
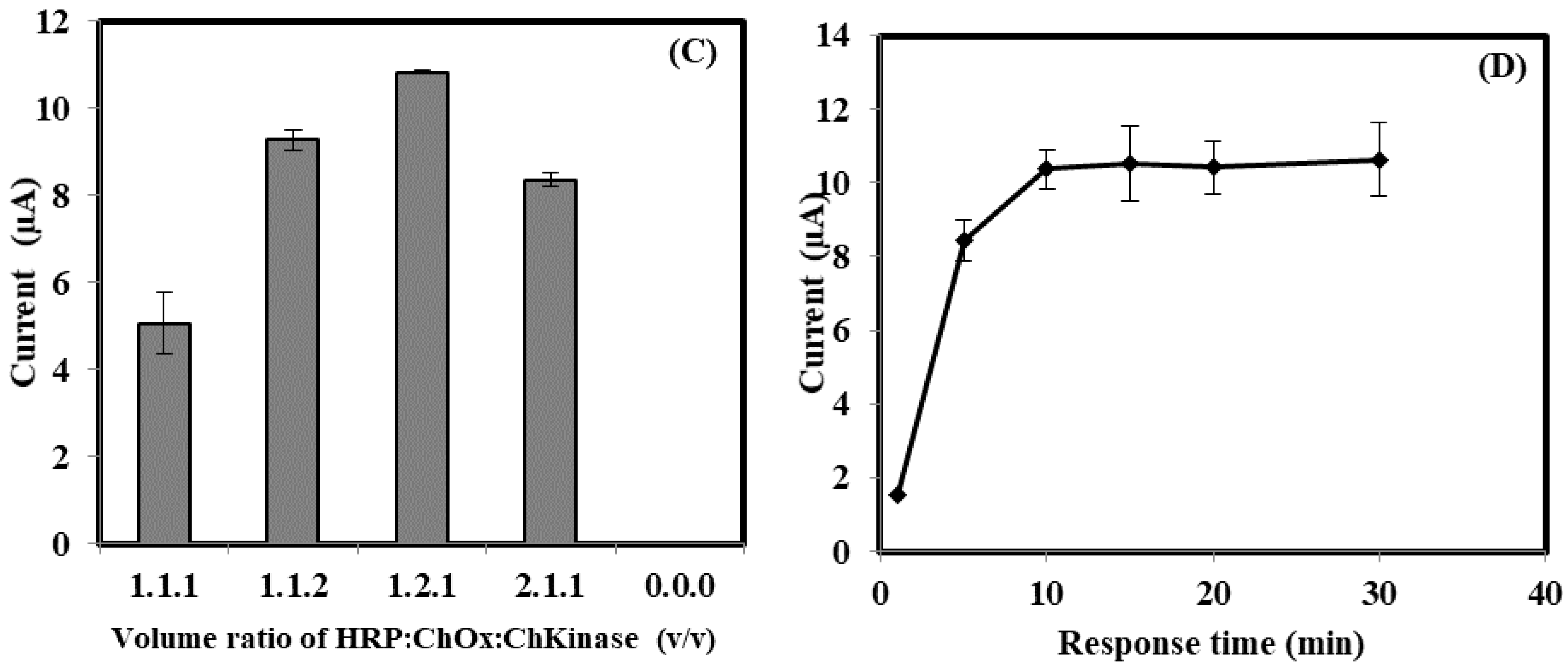
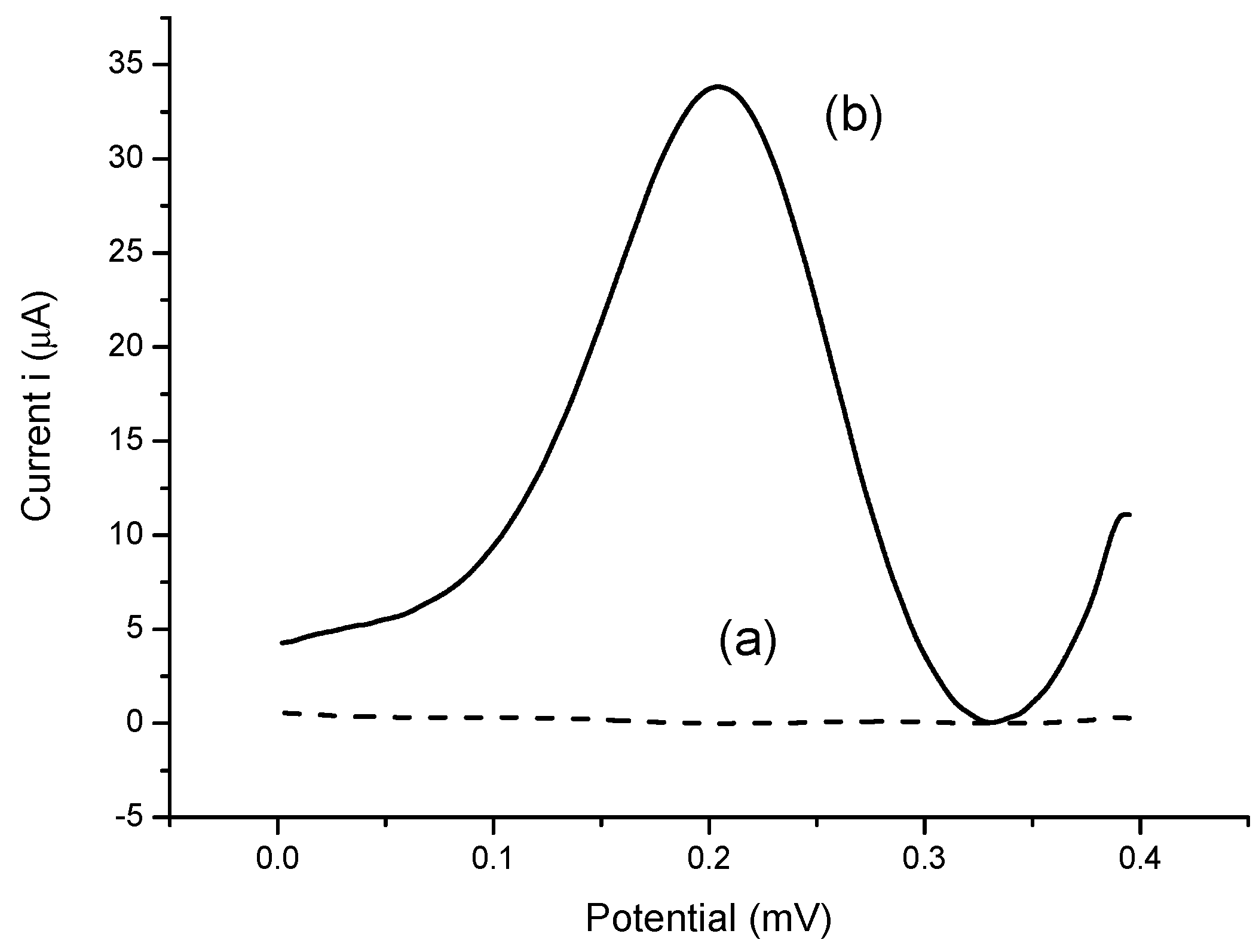
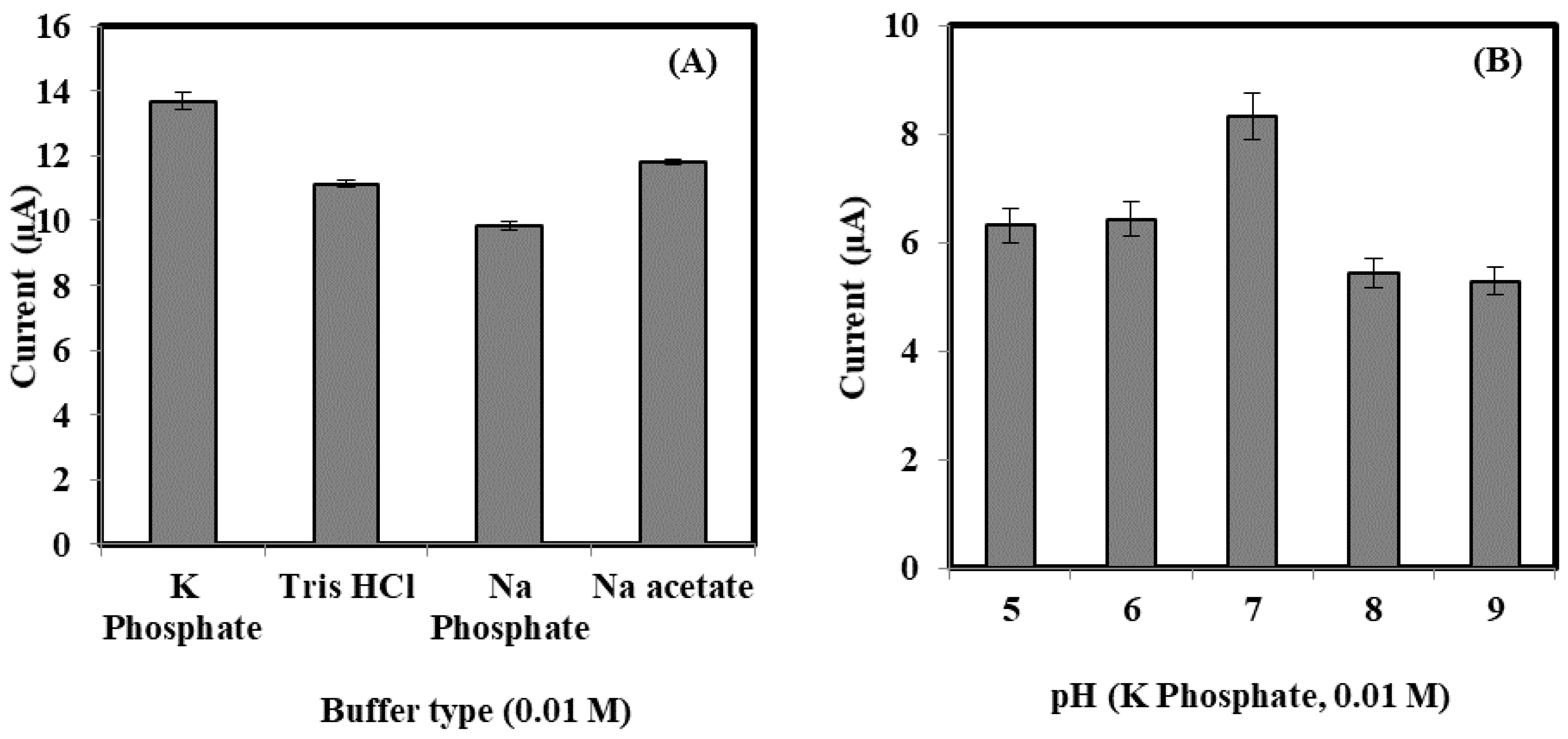
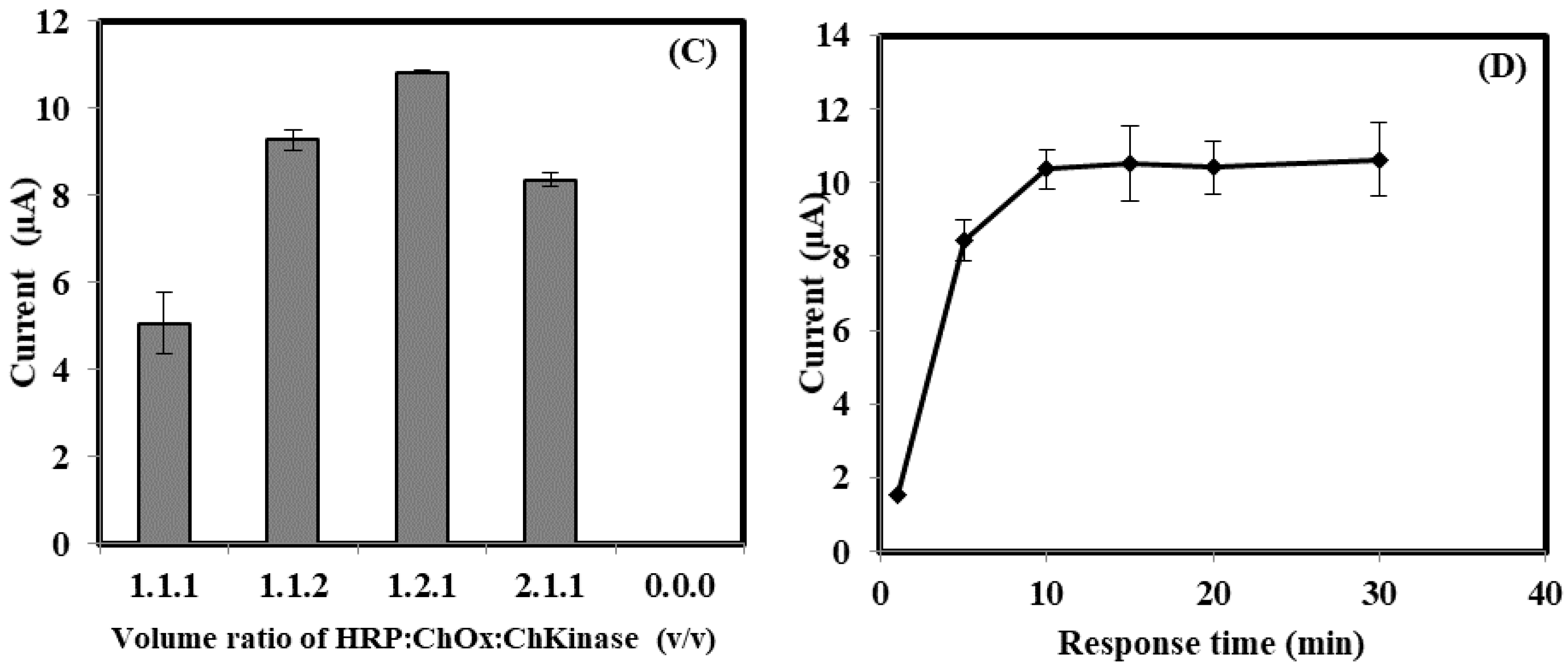
3.2. The Optimization of Bacterial Sepsis Biosensor
3.3. Selectivity Study
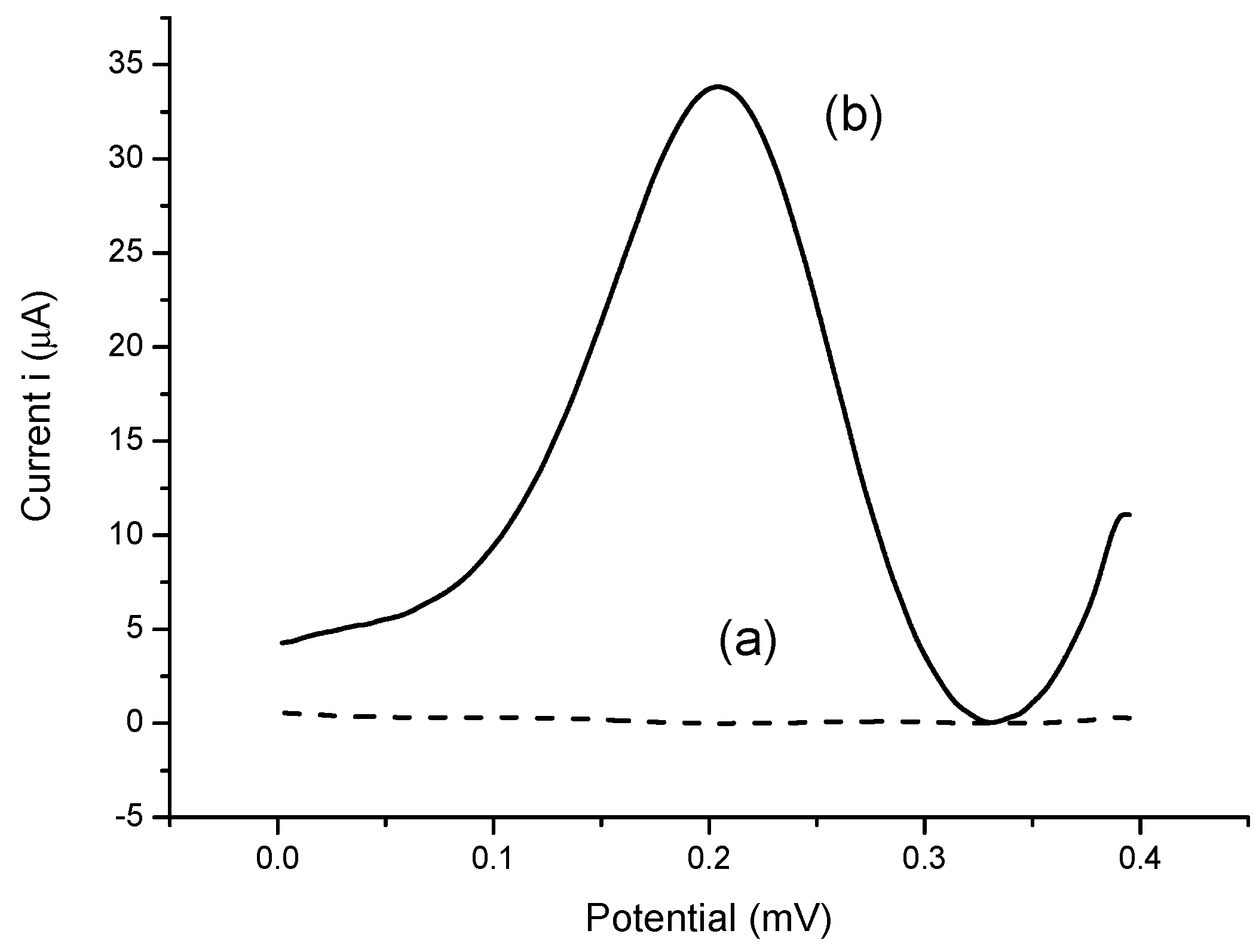
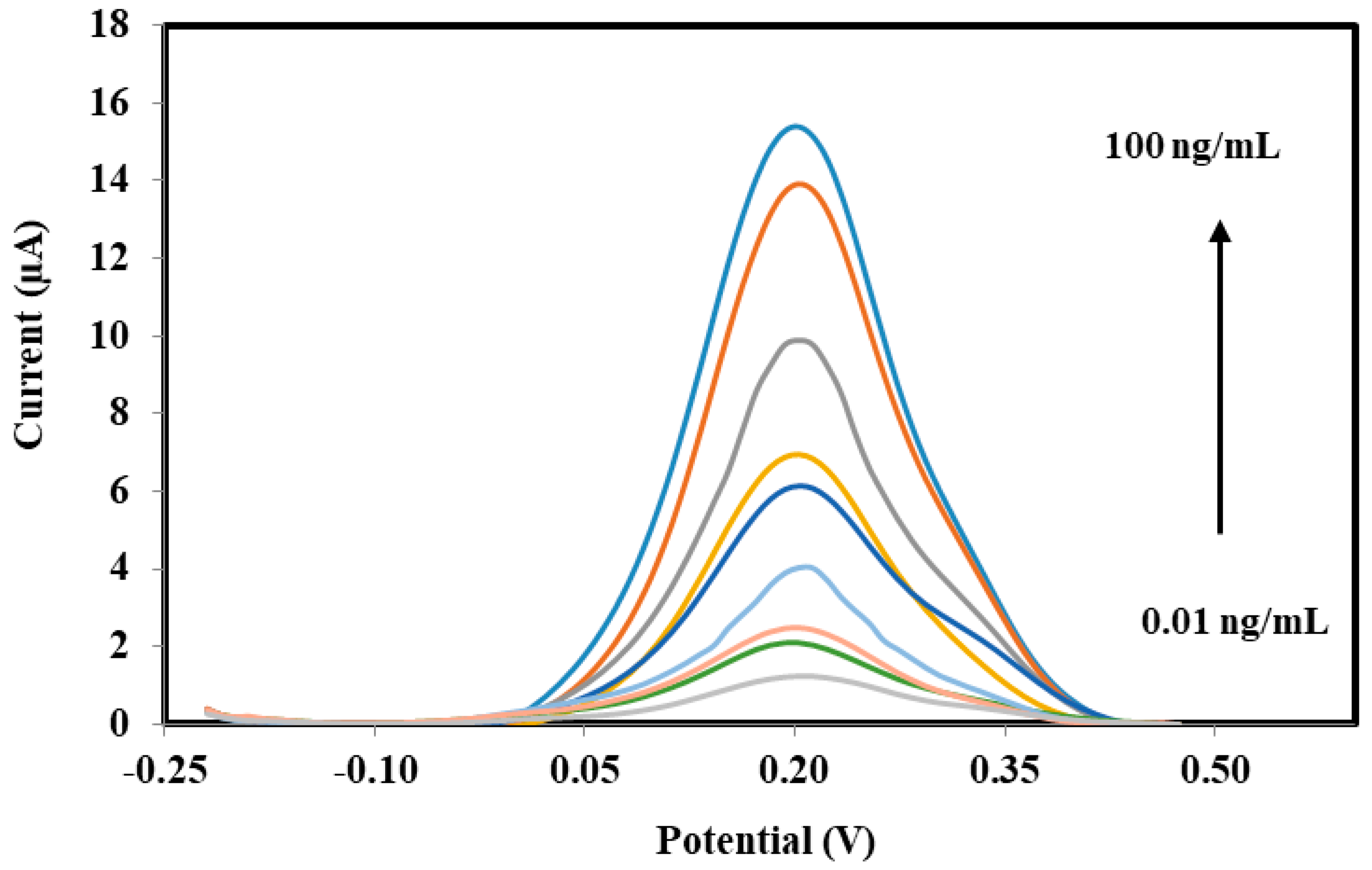
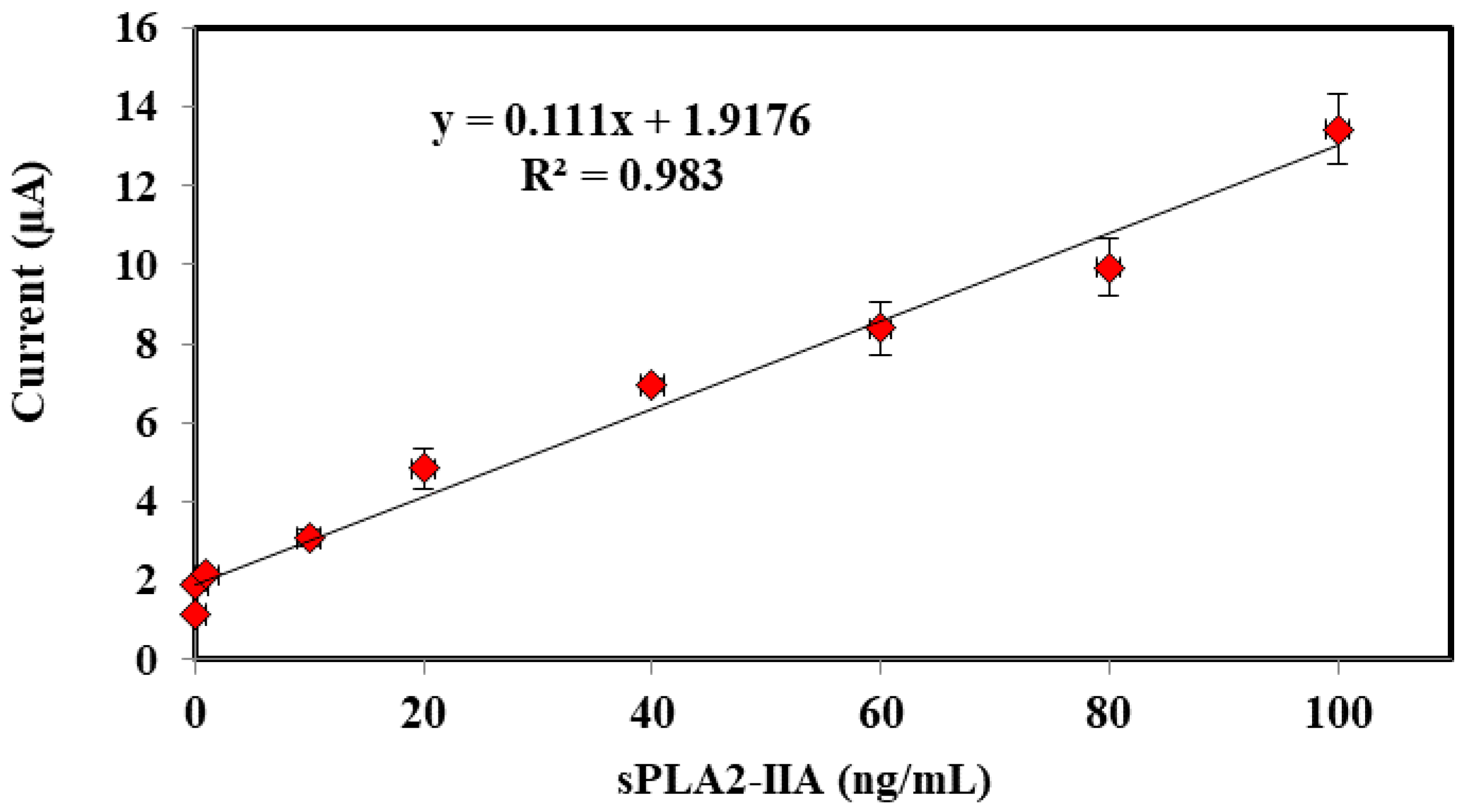
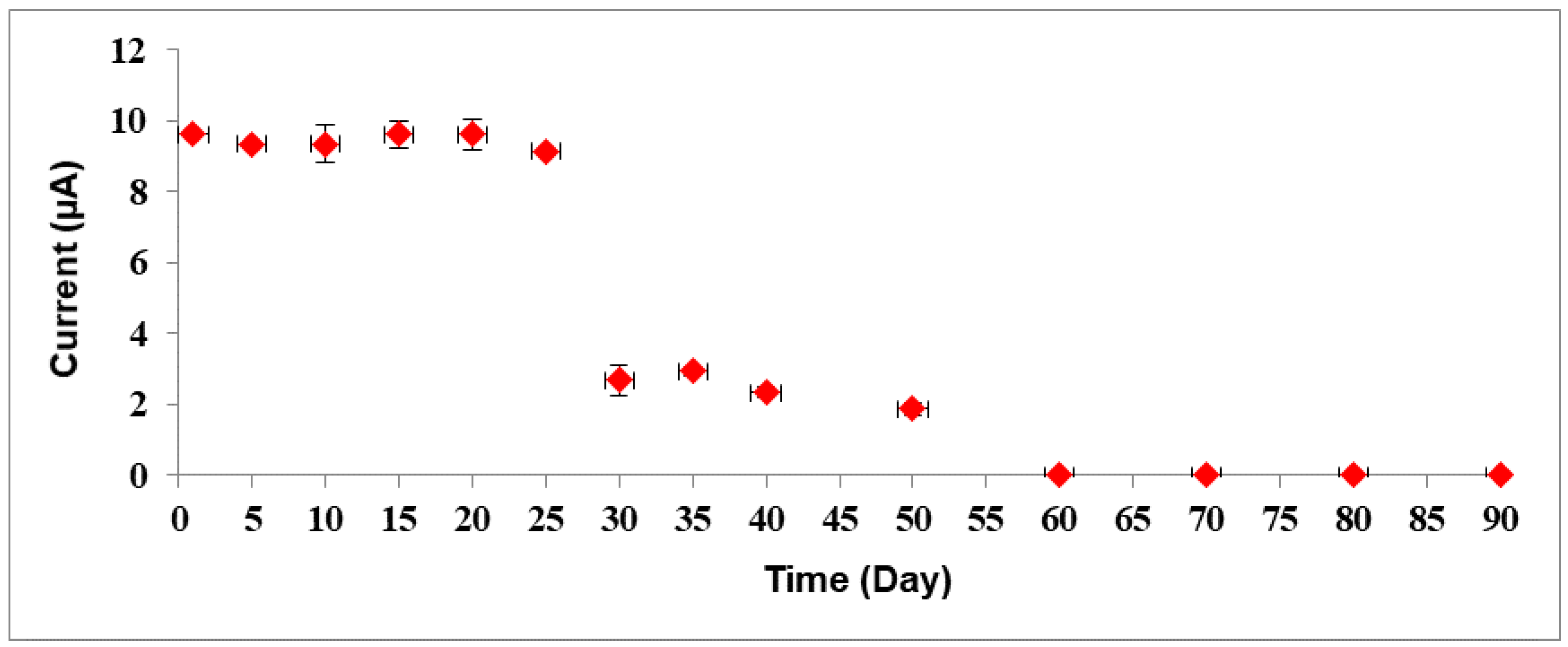
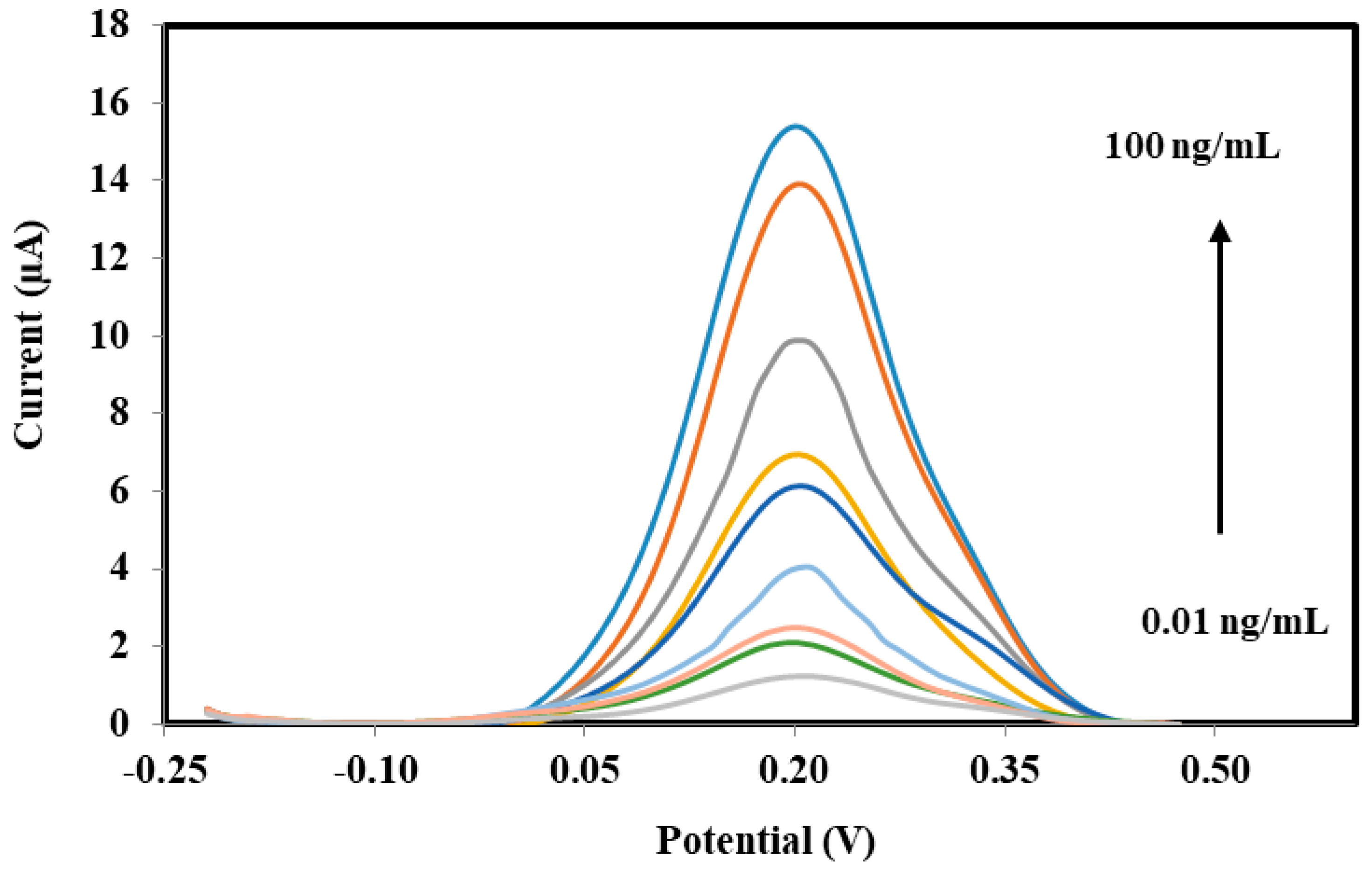
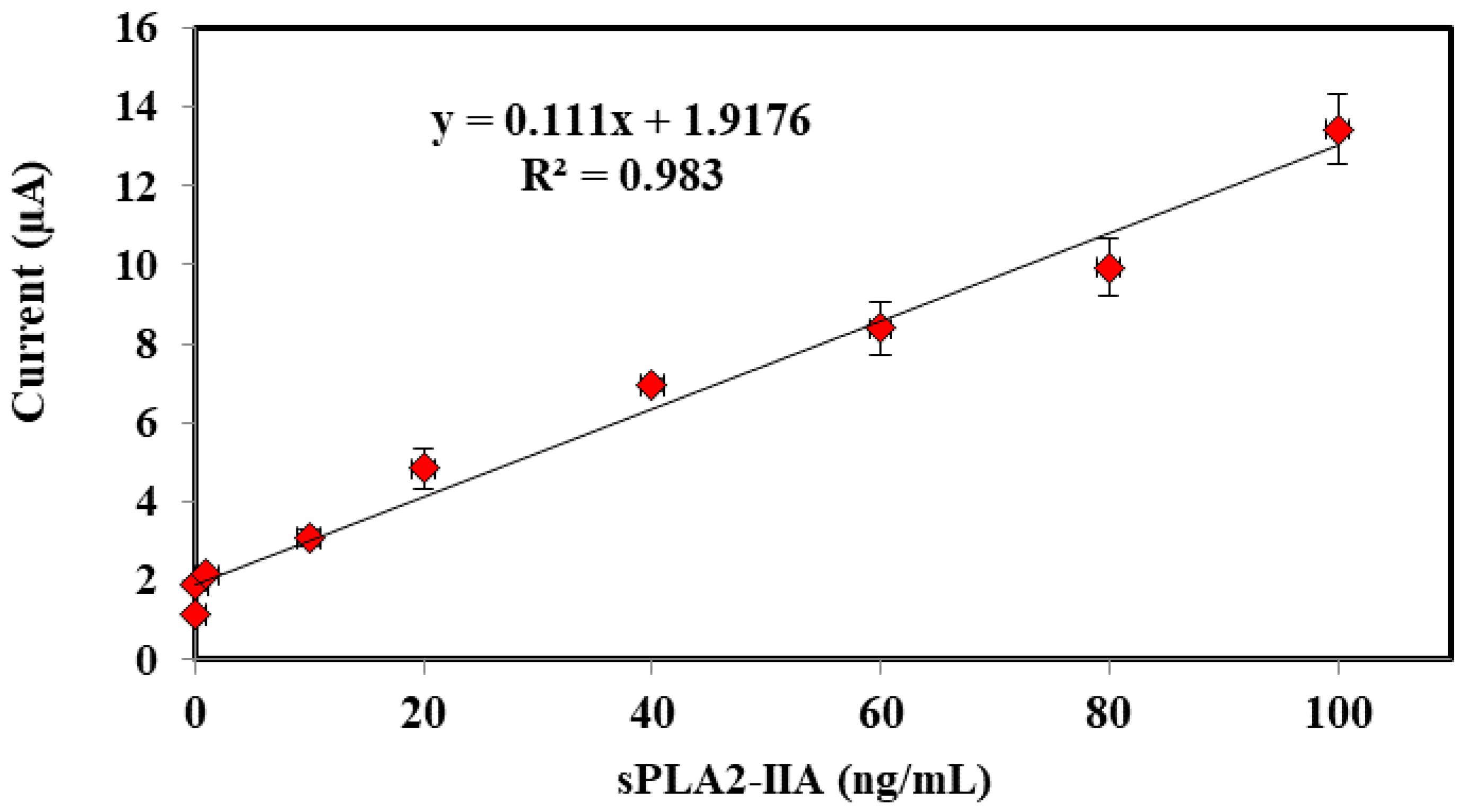
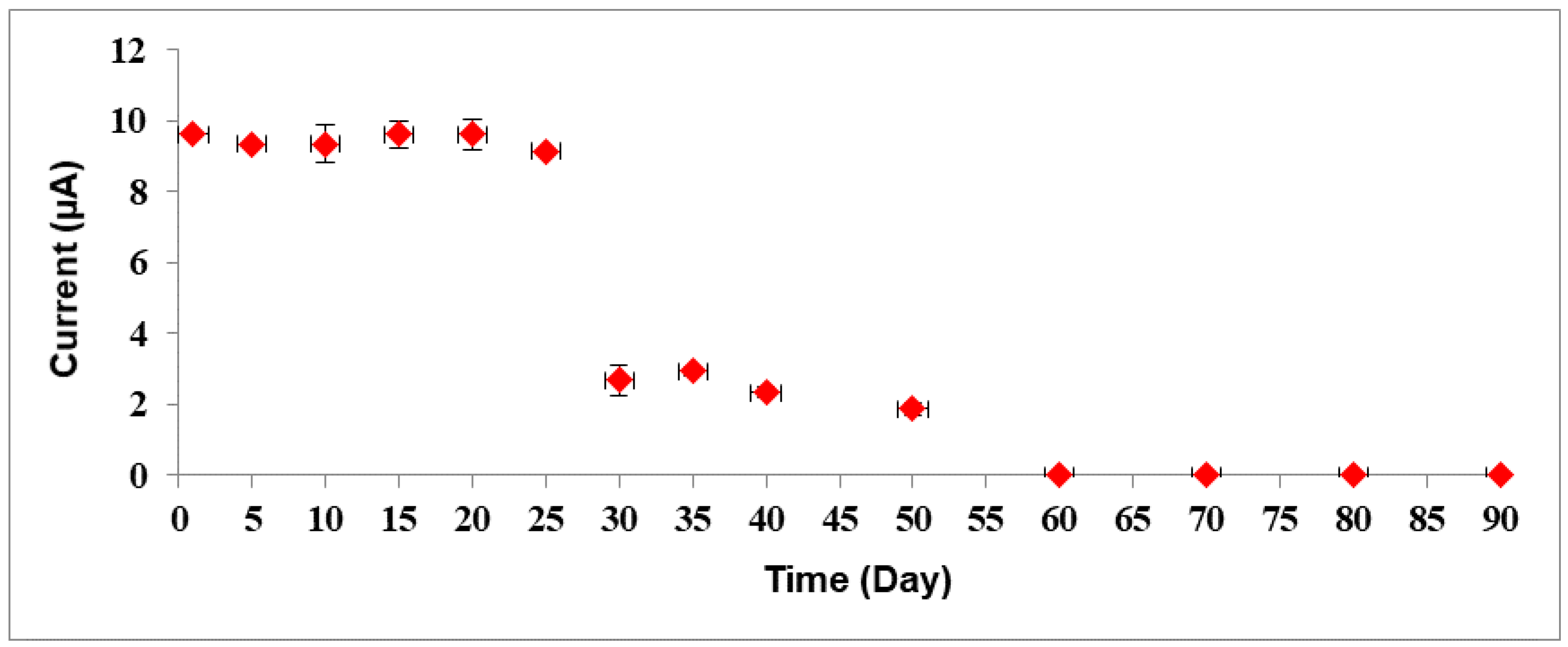
3.4. Performances of Bacterial Sepsis Biosensor
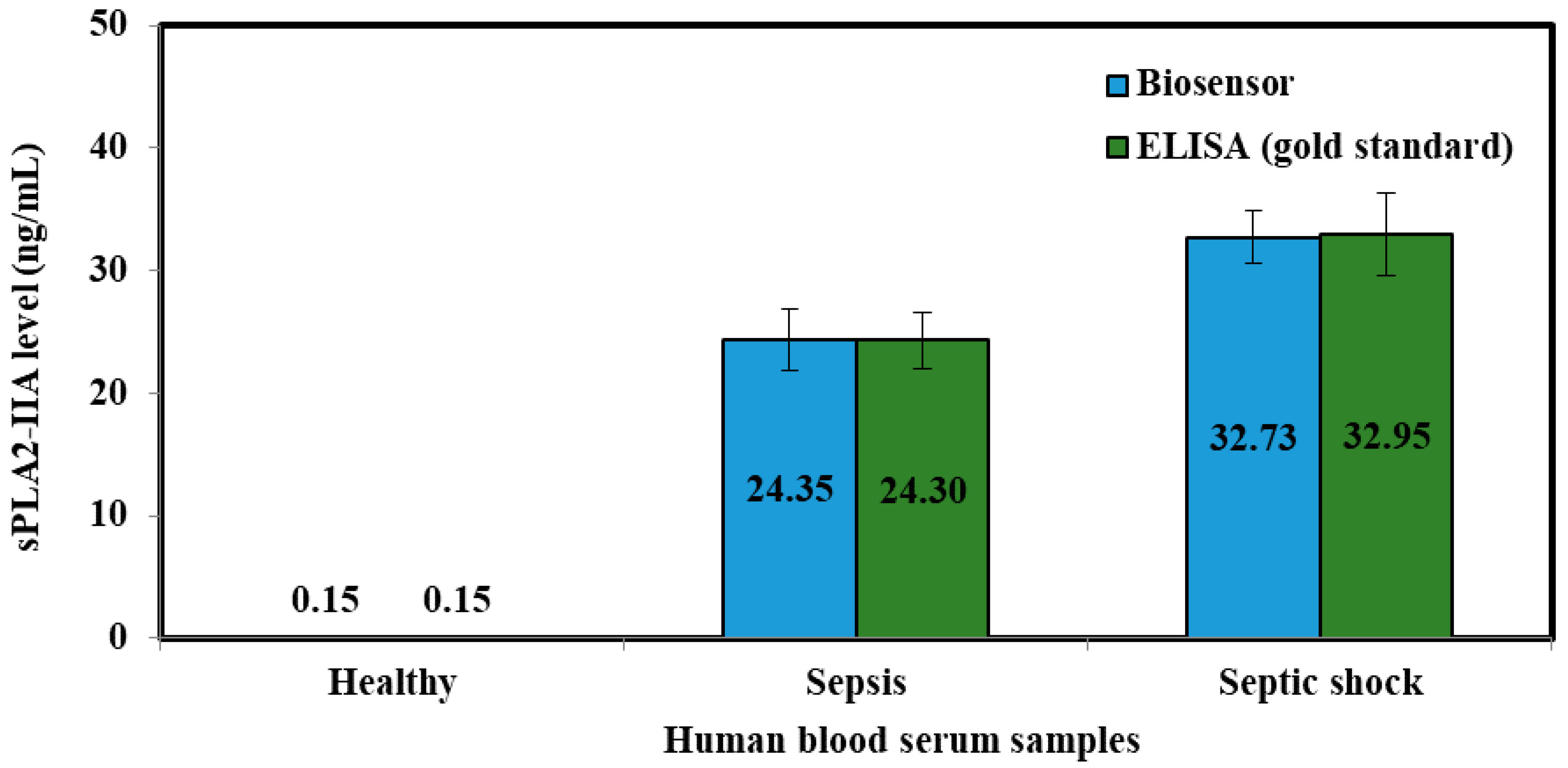
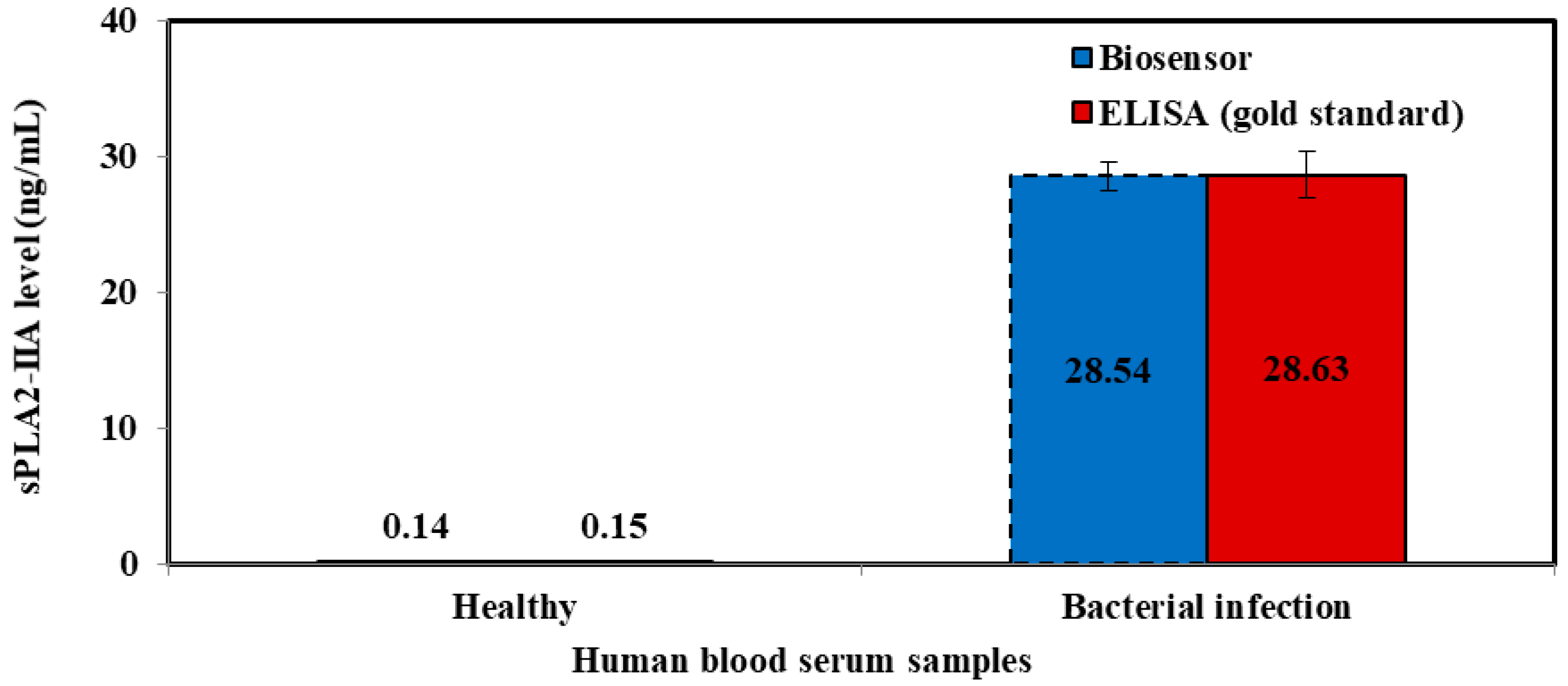
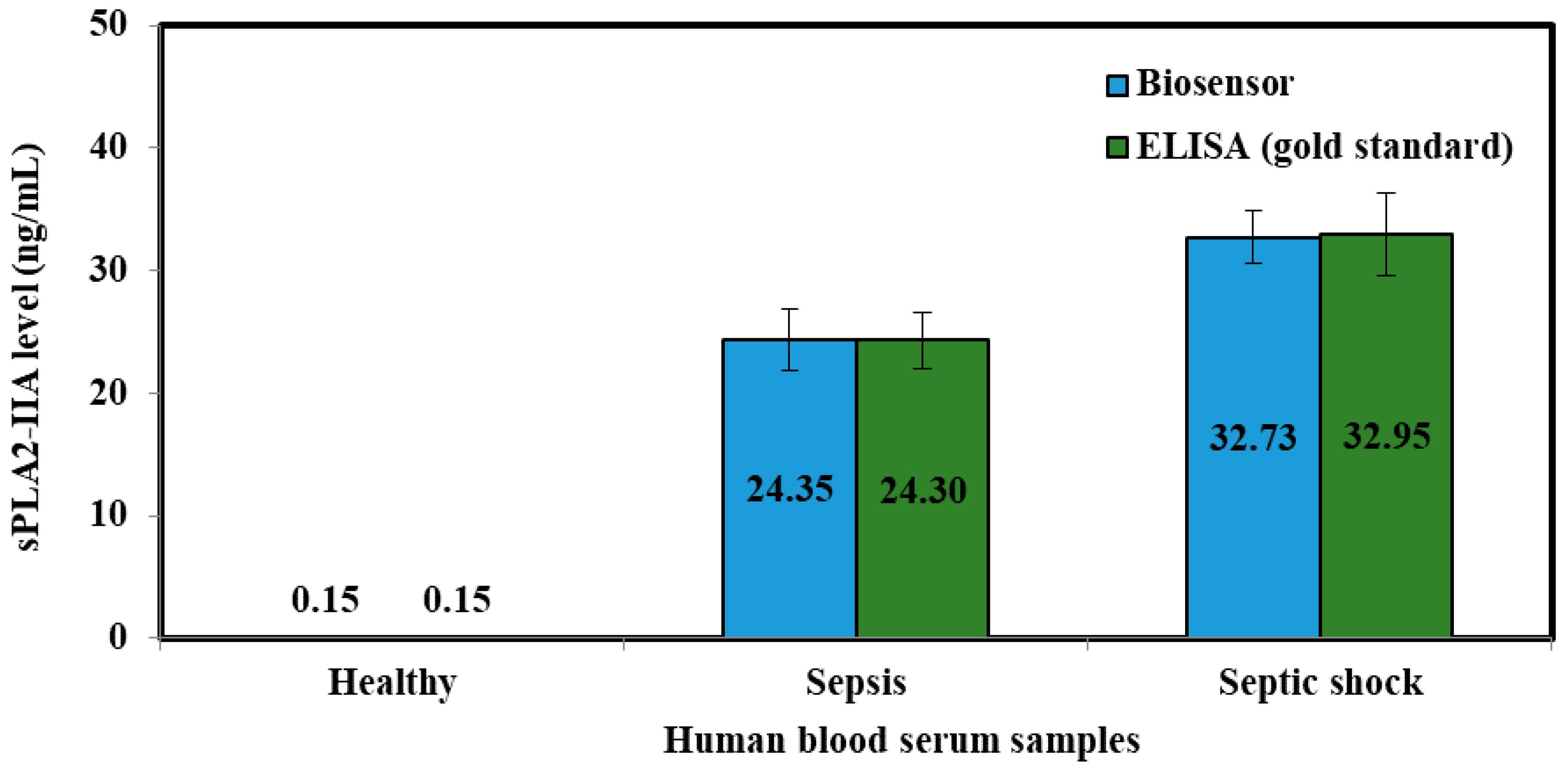
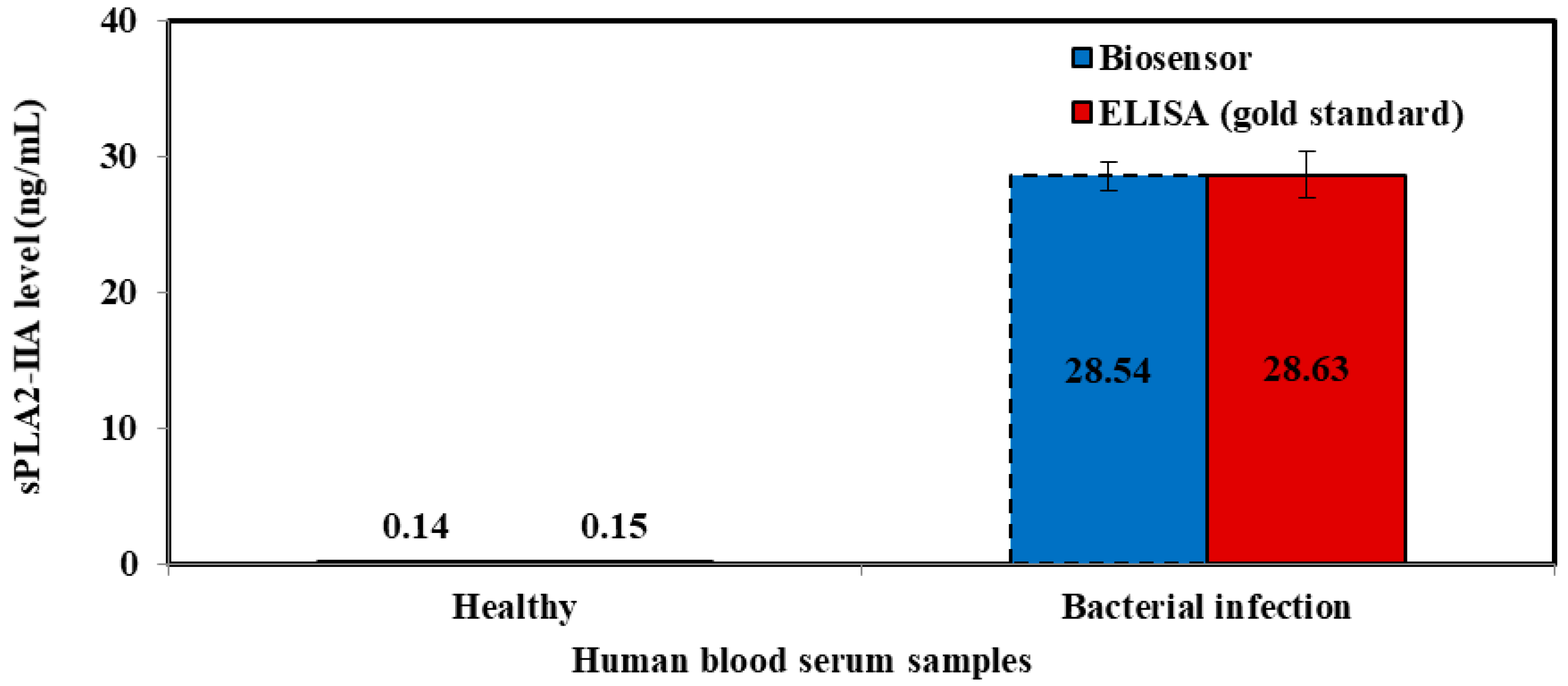
3.5. Validation of the Sepsis Biosensor Using Human Serum Smples
4. Conclusions
5. Patents
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bone, R.C.; Balk, R.A.; Cerra, F.B.; Dellinger, R.P.; Fein, A.M.; Knaus, W.A.; Schein, R.M.H.; Sibbald, W.J. Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. Chest 1992, 101, 1644–1655. [Google Scholar] [CrossRef] [PubMed]
- Davis, B.H. Improved diagnostic approaches to infection/sepsis detection. Expert Rev. Mol. Diagn. 2005, 5, 193–207. [Google Scholar] [CrossRef] [PubMed]
- Pierrakos, C.; Vincent, J.-L. Sepsis biomarkers: A review. Crit. Care 2010, 14, R15. [Google Scholar] [CrossRef] [PubMed]
- Rintala, M.; Aittoniemi, J.; Laine, S.; Nevalainen, T.; Nikoskelainen, J. Early identification of bacteremia by biochemical markers of systemic inflammation. Scand. J. Clin. Lab. Investig. 2001, 61, 523–530. [Google Scholar] [CrossRef]
- Nuutila, J.; Hohenthal, U.; Laitinen, I.; Kotilainen, P.; Rajamäki, A.; Nikoskelainen, J.; Lilius, E.-M. Simultaneous quantitative analysis of FcγRI (CD64) expression on neutrophils and monocytes: A new, improved way to detect infections. J. Immunol. Methods 2007, 328, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Cardelli, P.; Ferraironi, M.; Amodeo, R.; Tabacco, F.; De Blasi, R.; Nicoletti, M.; Sessa, R.; Petrucca, A.; Costante, A.; Cipriani, P. Evaluation of neutrophil CD64 expression and procalcitonin as useful markers in early diagnosis of sepsis. Int. J. Immunopathol. Pharmacol. 2008, 21, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Nevalainen, T.J.; Eerola, L.I.; Rintala, E.; Laine, V.J.O.; Lambeau, G.; Gelb, M.H. Time-resolved fluoroimmunoassays of the complete set of secreted phospholipases A2 in human serum. Biochim. Biophys. Acta Mol. Cell Bol. Lipids 2005, 1733, 210–223. [Google Scholar] [CrossRef] [PubMed]
- Guidet, B.; Piot, O.; Maury, E.; Offenstadt, G.; Masliah, J.; Bereziat, G.; Barakett, V. Secretory non-pancreatic phopholipase A2 in severe sepsis: Relation to endotoxin, cytokines and thromboxane B2. Infection 1996, 24, 103–108. [Google Scholar] [CrossRef] [PubMed]
- Nyman, K.M.; Uhl, W.; Forsström, J.; Büchler, M.; Beger, H.G.; Nevalainen, T.J. Serum phospholipase A2 in patients with multiple organ failure. J. Surg. Res. 1996, 60, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Rintala, E.M.; Nevalainen, T.J. Group II phospholipase A2 in sera of febrile patients with microbiologically or clinically documented infections. Clin. Infect. Dis. 1993, 17, 864–870. [Google Scholar] [CrossRef] [PubMed]
- Rintala, E.; Pulkki, K.; Mertsola, J.; Nevalainen, T.; Nikoskelainen, J. Endotoxin, interleukin-6 and phospholipase-A2 as markers of sepsis in patients with hematological malignancies. Scand. J. Infect. Dis. 1994, 27, 39–43. [Google Scholar]
- Hietaranta, A.; Kemppainen, E.; Puolakkainen, P.; Sainio, V.; Haapiainen, R.; Peuravuori, H.; Kivilaakso, E.; Nevalainen, T. Extracellular phospholipases A2 in relation to systemic inflammatory response syndrome (SIRS) and systemic complications in severe acute pancreatitis. Pancreas 1999, 18, 385–391. [Google Scholar] [CrossRef] [PubMed]
- Faix, J.D. Biomarkers of sepsis. Crit. Rev. Clin. Lab. Sci. 2013, 50, 23–36. [Google Scholar] [CrossRef] [PubMed]
- Tan, T.L.; Goh, Y.Y. The role of group IIA secretory phospholipase A2 (sPLA2-IIA) as a biomarker for the diagnosis of sepsis and bacterial infection in adults—A systematic review. PLoS ONE 2017, 12, e0180554. [Google Scholar] [CrossRef] [PubMed]
- Tan, T.L.; Ahmad, N.S.; Nasuruddin, D.N.; Ithnin, A.; Tajul Arifin, K.; Zaini, I.Z.; Wan Ngah, W.Z. CD64 and Group II Secretory Phospholipase A2 (sPLA2-IIA) as Biomarkers for Distinguishing Adult Sepsis and Bacterial Infections in the Emergency Department. PLoS ONE 2016, 11, e0152065. [Google Scholar] [CrossRef] [PubMed]
- Rosmawani, M.; Ahmad, M.; Heng, L.Y. Amperometric capsaicin biosensor based on covalent immobilization of horseradish peroxidase (HRP) on acrylic microspheres for chilli hotness determination. Sens. Actuators B Chem. 2017, 241, 174–181. [Google Scholar]
- Ling, Y.P.; Heng, L.Y. A potentiometric formaldehyde biosensor based on immobilization of alcohol oxidase on acryloxysuccinimide-modified acrylic microspheres. Sensors 2010, 10, 9963–9981. [Google Scholar] [CrossRef] [PubMed]
- Ulianas, A.; Heng, L.Y.; Ahmad, M. Biosensor for Urea from Succinimide-Modified Acrylic Microspheres Based on Reflectance Transduction. Sensors 2011, 11, 8323–8338. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.Q.; Guan, Y.P.; Liu, H.Z.; Ma, Z.Y.; Yang, Y.; Wu, X.B. Preparation and characterization of magnetic polymer nanospheres with high protein binding capacity. J. Magn. Magn. Mater. 2005, 293, 111–118. [Google Scholar] [CrossRef]
- Ulianas, A.; Heng, L.Y.; Ahmad, M.; Lau, H.-Y.; Ishak, Z.; Ling, T.L. A regenerable screen-printed DNA biosensor based on acrylic microsphere-gold nanoparticle composite for genetically modified soybean determination. Sens. Actuators B Chem. 2014, 190, 694–701. [Google Scholar] [CrossRef]
- Hirst, N.A.; Hazelwood, L.D.; Jayne, D.G.; Millner, P.A. An amperometric lactate biosensor using H2O2 reduction via a Prussian Blue impregnated poly(ethyleneimine) surface on screen printed carbon electrodes to detect anastomotic leak and sepsis. Sens. Actuators B Chem. 2013, 186, 674–680. [Google Scholar] [CrossRef]
- Kafi, A.K.; Lee, D.Y.; Park, S.H.; Kwon, Y.S. A hydrogen peroxide biosensor based on peroxidase activity of hemoglobin in polymeric film. J. Nanosci. Nanotechnol. 2007, 7, 4005–4008. [Google Scholar] [CrossRef] [PubMed]
- Tan, X.C.; Zhang, J.L.; Tan, S.W.; Zhao, D.D.; Huang, Z.W.; Mi, Y.; Huang, Z.Y. Amperometric hydrogen peroxide biosensor based on immobilization of hemoglobin on a glassy carbon electrode modified with Fe3O4/chitosan core-shell microspheres. Sensors 2009, 9, 6185–6199. [Google Scholar] [CrossRef] [PubMed]
- Diaz, A.N.; Peinado, M.C.R.; Minguez, M.C.T. Sol-gel horseradish peroxidase biosensor for hydrogen peroxide detection by chemiluminescence. Anal. Chim. Acta 1998, 363, 221–227. [Google Scholar] [CrossRef]
- Narang, J.; Minakshi; Bhambi, M.; Pundir, C.S. Fabrication of an amperometric triglyceride biosensor baed on PVC membrane. Anal. Lett. 2010, 43, 2009–2019. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Parameters | Value Range | Optimal Value |
|---|---|---|---|
| 1 | Amount of AuNPs (mg) | 0.03–0.1 | 0.05 |
| 2 | Amount of ACMS (mg) | 0.02–0.2 | 0.04 |
| Metabolite Compounds | DPV Peak Current (μA) | Sepsis Biosensor (%) | |
|---|---|---|---|
| 80 ng/mL | 40 ng/mL | ||
| sPLA2-IIA | 16.11 ± 1.02 | 100 | 100 |
| Ascorbic acid | 3.16 ± 0.41 | 19.6 | 18.4 |
| Urea | 1.83 ± 0.07 | 11.4 | 10 |
| Sucrose | 1.58 ± 0.02 | 9.8 | 9.5 |
| Glucose | 1.24 ± 0.03 | 7.7 | 6.5 |
| Citric acid | 0.93 ± 0.02 | 5.8 | 4.9 |
| Conc. (sPLA2-IIA) | Mean ± SD (Current, μA) | RSD (%, n = 3) |
|---|---|---|
| 0.01 | 1.14 ± 0.053 | 4.673 |
| 0.10 | 1.88 ± 0.157 | 8.367 |
| 1 | 2.14 ± 0.049 | 2.287 |
| 10 | 3.09 ± 0.211 | 6.851 |
| 20 | 4.84 ± 0.509 | 10.525 |
| 40 | 6.95 ± 0.146 | 2.101 |
| 60 | 8.38 ± 0.684 | 8.162 |
| 80 | 9.94 ± 0.738 | 7.424 |
| 100 | 13.43 ± 0.882 | 6.567 |
| Samples | Mean ± SD (Current, μA) | RSD (%, n = 3) | Plotted (ng/mL) | Elisa Method (ng/mL) |
|---|---|---|---|---|
| Healthy | 1.934 ± 0.189 | 9.786 | 0.145 | 0.148 |
| Sepsis | 4.620 ± 0.465 | 10.061 | 24.349 | 24.30 |
| Septic shock | 5.551 ± 0.521 | 9.379 | 32.731 | 32.95 |
| Samples | sPLA2-IIA Level (ng/mL) | |
|---|---|---|
| Mean ± SD | RSD (%, n = 3) | |
| Healthy | 0.145 ± 0.009 | 6.174 |
| Sepsis | 24.35 ± 2.457 | 10.090 |
| Septic shock | 32.73 ± 2.102 | 6.423 |
| Samples | Biosensor Method | ELISA Method | t Test | ||
|---|---|---|---|---|---|
| Current (μA) | RSD (%, n = 3) | Human sPLA2-IIA (ng/mL) | Human sPLA2-IIA (ng/mL) | ||
| Healthy | 1.93 ± 0.19 | 9.78 | 0.145 ± 0.01 | 0.148 ± 0.01 | 0.117 |
| Sepsis | 10.06 ± 0.37 | 7.90 | 24.349 ± 2.46 | 24.303 ± 2.31 | 0.018 |
| Septic shock | 5.55 ± 0.42 | 7.57 | 32.732 ± 2.10 | 32.950 ± 3.37 | 0.091 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nik Mansor, N.N.; Leong, T.T.; Safitri, E.; Futra, D.; Ahmad, N.S.; Nasuruddin, D.N.; Itnin, A.; Zaini, I.Z.; Arifin, K.T.; Heng, L.Y.; et al. An Amperometric Biosensor for the Determination of Bacterial Sepsis Biomarker, Secretory Phospholipase Group 2-IIA Using a Tri-Enzyme System. Sensors 2018, 18, 686. https://doi.org/10.3390/s18030686
Nik Mansor NN, Leong TT, Safitri E, Futra D, Ahmad NS, Nasuruddin DN, Itnin A, Zaini IZ, Arifin KT, Heng LY, et al. An Amperometric Biosensor for the Determination of Bacterial Sepsis Biomarker, Secretory Phospholipase Group 2-IIA Using a Tri-Enzyme System. Sensors. 2018; 18(3):686. https://doi.org/10.3390/s18030686
Chicago/Turabian StyleNik Mansor, Nik Nurhanan, Tan Toh Leong, Eka Safitri, Dedi Futra, Nurul Saadah Ahmad, Dian Nasriana Nasuruddin, Azlin Itnin, Ida Zarina Zaini, Khaizurin Tajul Arifin, Lee Yook Heng, and et al. 2018. "An Amperometric Biosensor for the Determination of Bacterial Sepsis Biomarker, Secretory Phospholipase Group 2-IIA Using a Tri-Enzyme System" Sensors 18, no. 3: 686. https://doi.org/10.3390/s18030686
APA StyleNik Mansor, N. N., Leong, T. T., Safitri, E., Futra, D., Ahmad, N. S., Nasuruddin, D. N., Itnin, A., Zaini, I. Z., Arifin, K. T., Heng, L. Y., & Hassan, N. I. (2018). An Amperometric Biosensor for the Determination of Bacterial Sepsis Biomarker, Secretory Phospholipase Group 2-IIA Using a Tri-Enzyme System. Sensors, 18(3), 686. https://doi.org/10.3390/s18030686