
Analytical Protein Microarrays: Advancements Towards Clinical Applications


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. General Background
3. Supports and Immobilization Strategies
3.1. Pros and Cons of Oriented Antibody Immobilization
3.2. Surface Chemistries for Small Molecules
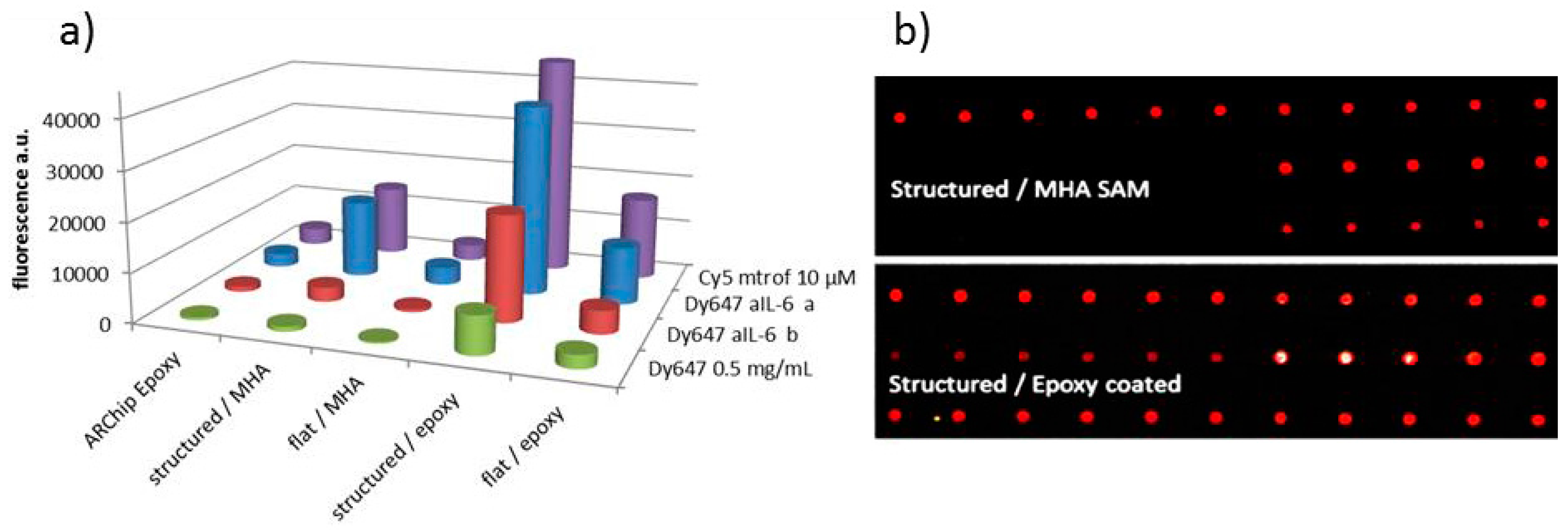
3.3. Coating of Substrates
4. Biological and Biomimetic Recognition Elements (BREs) in Immunoanalytical Microarrays
4.1. Antibodies
4.2. Peptides
4.3. Aptamers
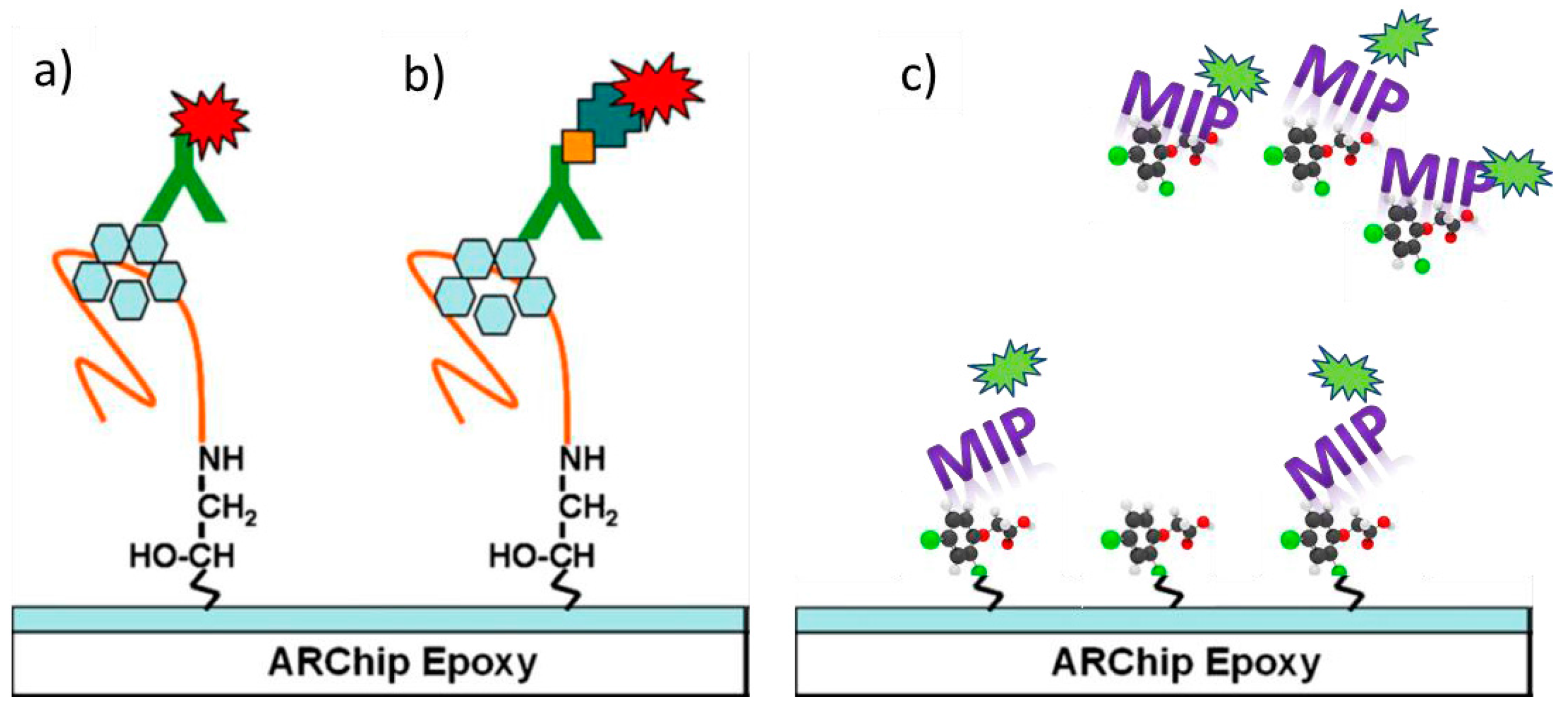
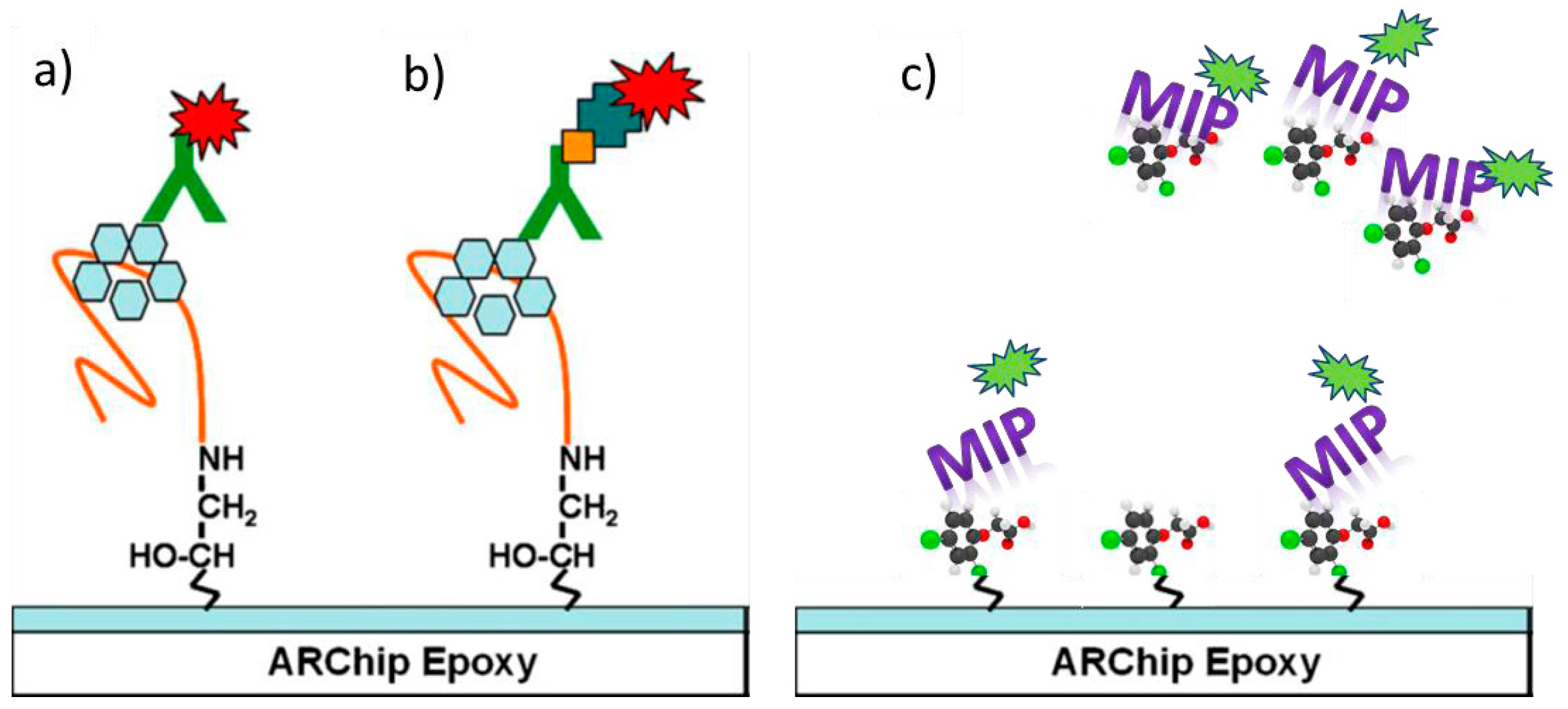
4.4. Molecularly Imprinted Polymers (MIPs)
5. Up-to-Date Patterning of BREs
5.1. Non-Contact Printing
5.2. Contact Printing
5.3. µ-Contact Printing (µCP)
5.4. Nanobiolithography
6. Sample Preparation
7. On-Chip Immunoassays
7.1. Platforms: Slides, Micro- or Nano Well Chips/Plates
7.2. Assay Formats
7.3. Immuno-PCR
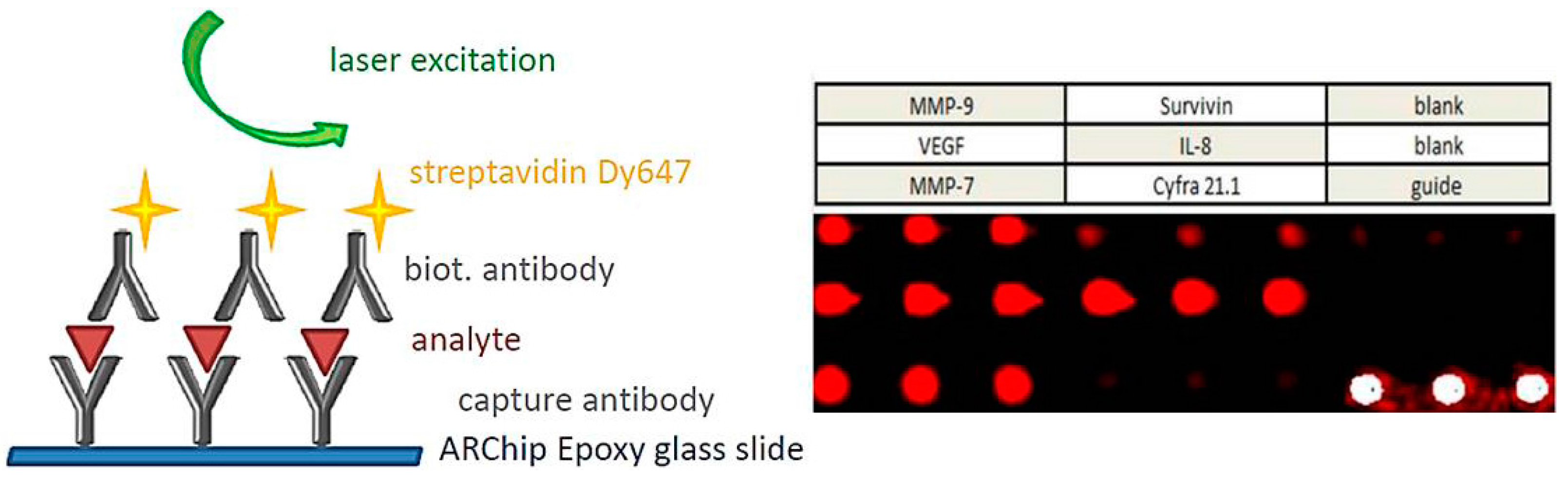
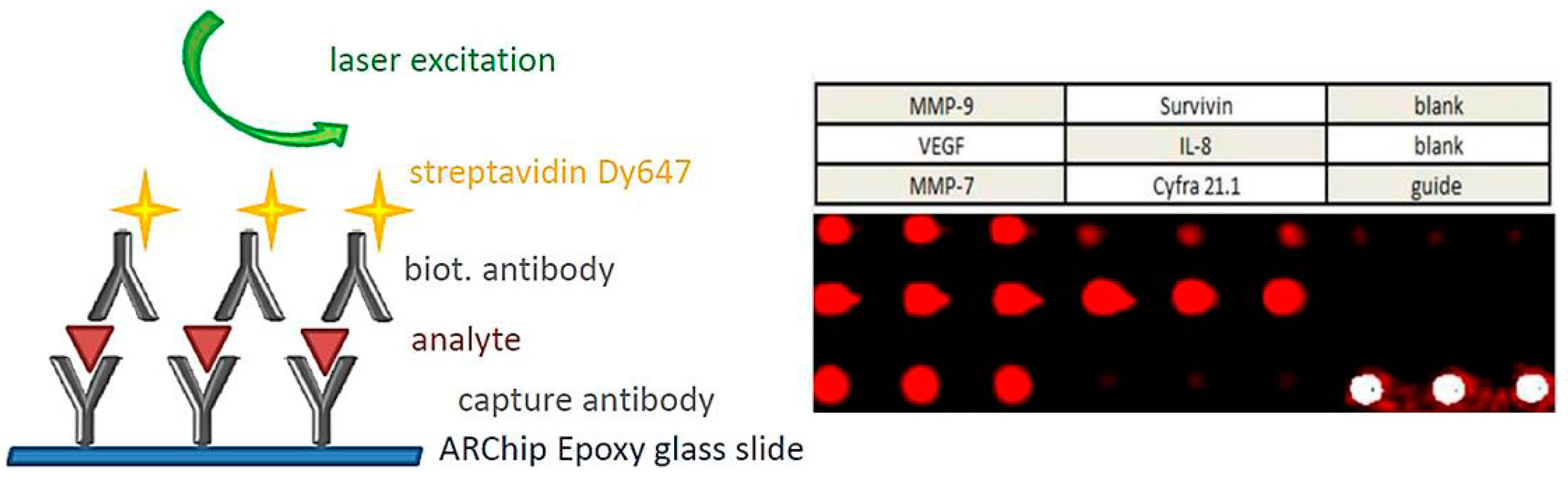
8. Signal Transduction and Read-Out
9. Signal Enhancement
- High density and accessibility of probes (i.e., immobilized biorecognition elements)
- High density of labels per binding event
- Enhanced intensity per fluorophore
10. Miniaturization
- -
- Higher spot density and consequently higher number of BREs on a given chip size;
- -
- Faster reaction kinetics and lower assay times;
- -
- Reduced consumption of reagents and most important of (patient) samples;
- -
- Small arrays avoid scanning and hence reduce size and costs of read-out instruments;
- -
- Reduced chip size is needed for integration into (portable) instruments.
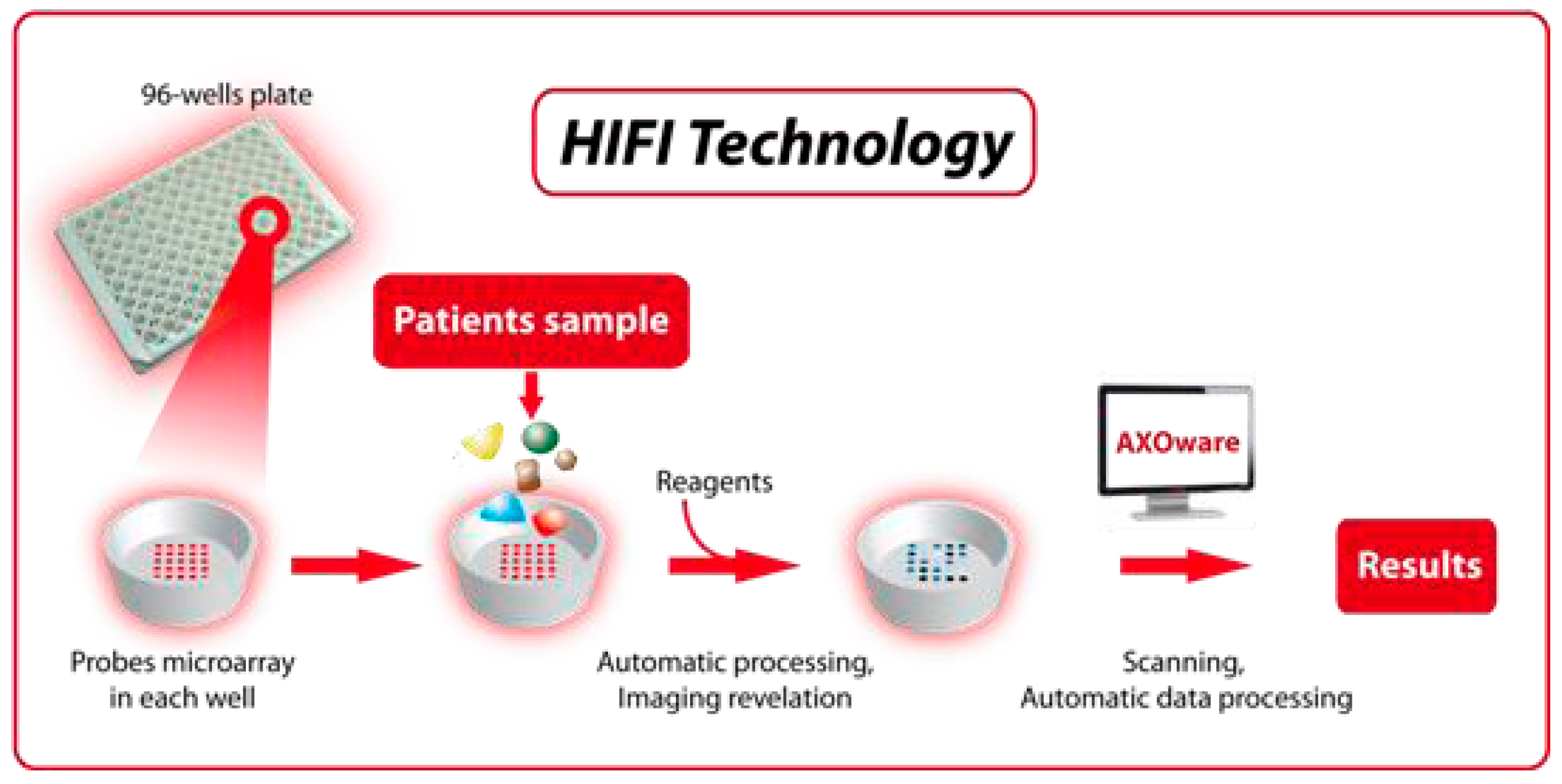
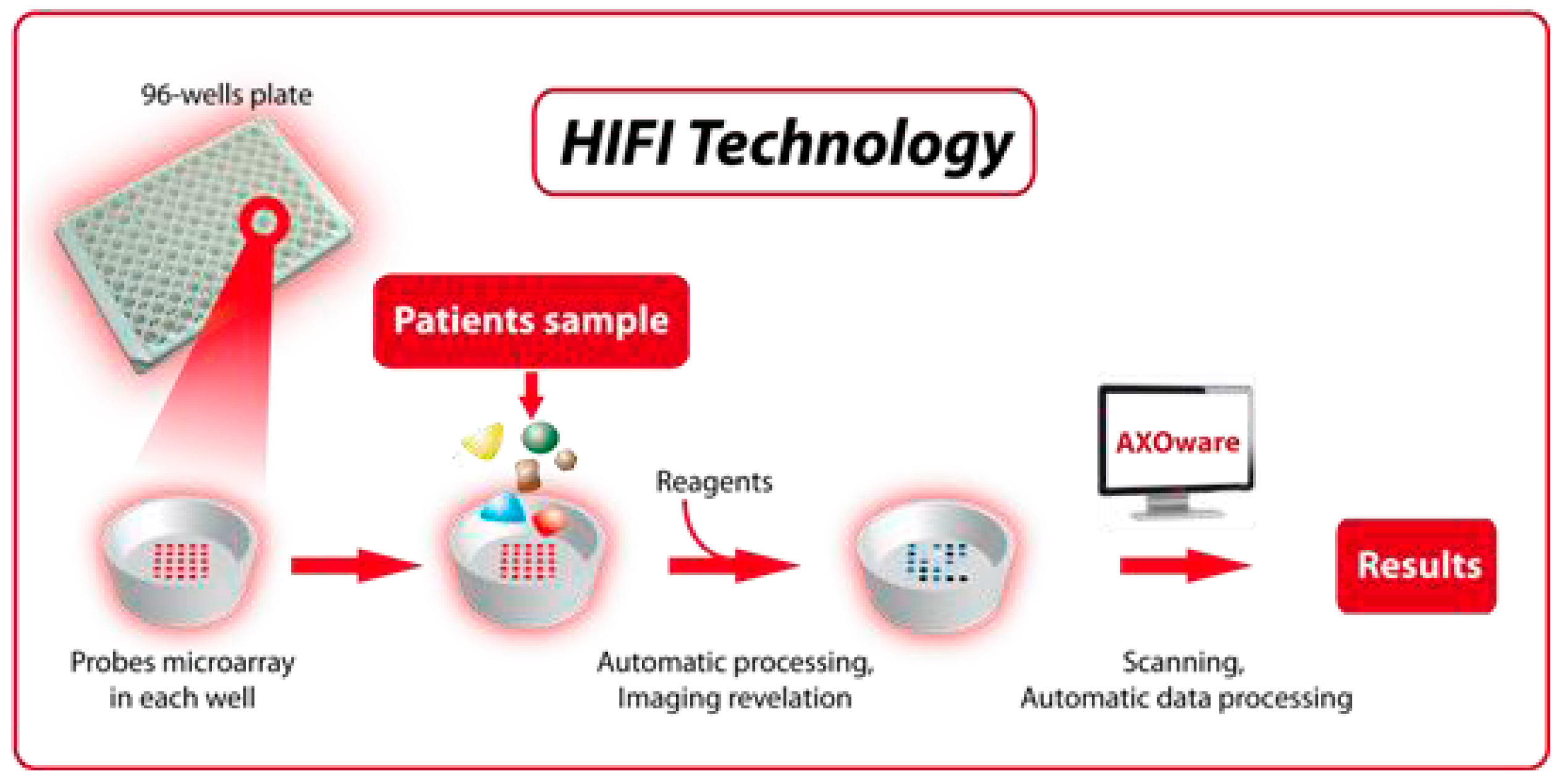
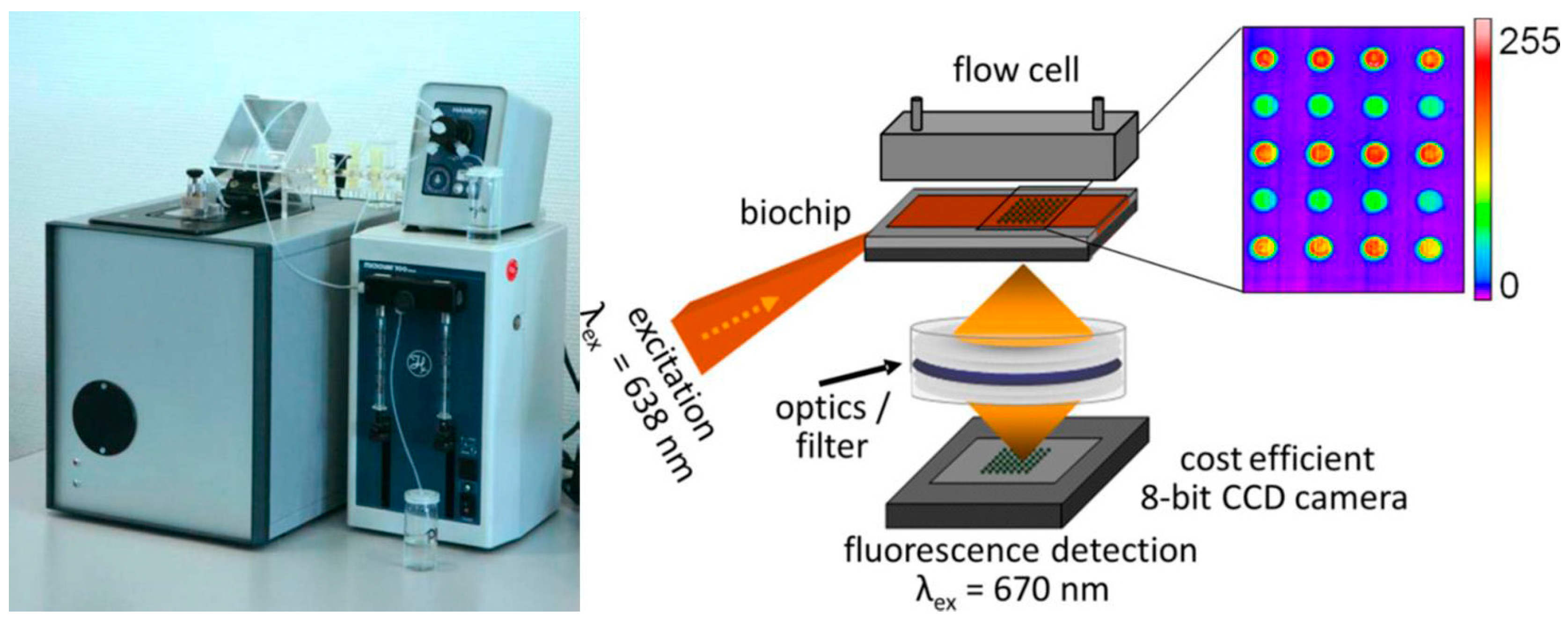
11. Automated Platforms
12. Conclusions
Acknowledgments
Conflicts of Interest
References
- Zhu, H.; Qian, J. Applications of Functional Protein Microarrays in Basic and Clinical Research. Adv. Genet. 2012, 79, 123–155. [Google Scholar] [PubMed]
- Schena, M. Protein Microarrays; Jones and Bartlett: Sudbury, MA, USA, 2005; p. 469. [Google Scholar]
- Preininger, C.; Sauer, U. Design, Quality Control and Normalization of Biosensor Chips. In Optical Sensors for Industrial and Environmental Applications, 1st ed.; Wolfbeis, O., Narayanaswamy, R., Eds.; Springer: Berlin, Germany, 2003; pp. 67–90. [Google Scholar]
- Preininger, C.; Sauer, U. Quality control of chip manufacture and chip analysis using epoxy-chips as a model. Sens. Actuators B Chem. 2003, 90, 98–103. [Google Scholar] [CrossRef]
- Sauer, U.; Preininger, C.; Hany-Schmatzberger, R. Quick and simple: Quality control of microarray data. Bioinformatics 2005, 21, 1572–1578. [Google Scholar] [CrossRef] [PubMed]
- Preininger, C.; Sauer, U.; Dayteg, J.; Pichler, R. Optimizing processing parameters for signal enhancement of oligonucleotide and protein arrays on ARChip Epoxy. Bioelectrochemistry 2005, 67, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Preininger, C.; Sauer, U.; Obersriebnig, S.; Trombitas, M. Signal enhancement of protein chips using 3D-materials for immobilization. Int. J. Environ. Anal. Chem. 2005, 85, 645–654. [Google Scholar] [CrossRef]
- Baker, M. Blame it on the antibodies. Nature 2015, 521, 274–276. [Google Scholar] [CrossRef] [PubMed]
- Baker, M. Antibody Anarchy: A call to order. Nature 2015, 527, 545–551. [Google Scholar] [CrossRef] [PubMed]
- Weller, M.G. Quality Issues of Research Antibodies. Anal. Chem. Insights 2016, 11, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Kost, G.; Ehrmeyer, S.; Chernow, B.; Winkelman, J.; Zaloga, G.; Dellinger, R.; Shirey, T. The laboratory–clinical interface—Point-of-care testing. Chest 1999, 115, 1140–1154. [Google Scholar] [CrossRef] [PubMed]
- Tsarfati-BarAd, I.; Sauer, U.; Preininger, C.; Gheber, L. Miniaturized protein arrays: Model and experiment. Biosens. Bioelectron. 2011, 26, 3774–3781. [Google Scholar] [CrossRef] [PubMed]
- Ekins, R.; Chu, F.; Biggart, E. Fluorescence spectroscopy and its application to a new generation of high sensitivity, multi-microspot, multianalyte, immunoassay. Clin. Chim. Acta 1990, 194, 91–114. [Google Scholar] [CrossRef]
- Ekins, R.; Chu, F. Multianalyte Microspot Immunoassay-Microanalytical “Compact Disk” of the Future. Clin. Chem. 1991, 37, 1955–1967. [Google Scholar] [PubMed]
- Nam, J.; Han, S.; Lee, K.; Liu, X.; Ratner, M.; Mirkin, C. Bioactive Protein Nanoarrays on Nickel Oxide Surfaces Formed by Dip-Pen Nanolithography. Angew. Chem. Int. Ed. 2004, 116, 1266–1269. [Google Scholar] [CrossRef]
- Chen, H.; Huang, J.; Lee, J.; Hwang, S.; Koh, K. Surface plasmon resonance spectroscopic characterization of antibody orientation and activity on the calixarene monolayer. Sens. Actuators B Chem. 2010, 147, 548–553. [Google Scholar] [CrossRef]
- Preininger, C.; Sauer, U.; Chouiki, M.; Schöftner, R. Nanostructures in protein chips: Effect of print buffer additive and wettability on immobilization and assay performance. Microelectron. Eng. 2011, 88, 1856–1859. [Google Scholar] [CrossRef]
- Zhu, H.; Snyder, M. Protein arrays and microarrays. Curr. Opin. Chem. Biol. 2001, 5, 40–45. [Google Scholar] [CrossRef]
- Arenkov, P.; Kukhtin, A.; Gemmell, A.; Voloshchuk, S.; Chupeeva, V.; Mirzabekov, A. Protein microchips: Use for immunoassay and enzymatic reactions. Anal. Biochem. 2000, 278, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Templin, M.F.; Stoll, D.; Schrenk, M.; Traub, P.C.; Vöhringer, C.F.; Joos, T.O. Protein Microarray Technology. Trends Biotechnol. 2002, 20, 160–166. [Google Scholar] [CrossRef]
- Haab, B.B.; Dunham, M.J.; Brown, P.O. Protein microarrays for highly parallel detection and quantitation of specific proteins and antibodies in complex solutions. Genome Biol. 2001, 2, research0004. [Google Scholar] [CrossRef] [PubMed]
- Guthy, D.A.; Voshol, H. Antibody-based arrays in disease proteomics. Antibody Technol. J. 2015, 5, 15–25. [Google Scholar]
- Kemmler, M.; Sauer, U.; Schleicher, E.; Preininger, C.; Brandenburg, A. Biochip point-of-care device for sepsis diagnostics. Sens. Actuators B Chem. 2014, 192, 205–215. [Google Scholar] [CrossRef]
- Domnanich, P.; Sauer, U.; Pultar, J.; Preininger, C. Protein microarray for the analysis of human melanoma biomarkers. Sens. Actuators B Chem. 2009, 139, 2–8. [Google Scholar] [CrossRef]
- Sereni, M.; Pierobon, M.; Angioli, R.; Petricoin, E.; Frederick, M. Reverse Phase Protein Microarrays and Their Utility in Drug Development. In Target Identification and Validation in Drug Discovery, 1st ed.; Moll, J., Colombo, R., Eds.; Humana Press: Mumbai, India, 2013; Volume 986, pp. 187–214. [Google Scholar]
- Tschmelak, J.; Proll, G.; Gauglitz, G. Optical biosensor for pharmaceuticals, antibiotics, hormones, endocrine disrupting chemicals and pesticides in water: Assay optimization process for estrone as example. Talanta 2005, 65, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Sapsford, K.; Ngundi, M.; Moore, M.; Lassman, M.; Shriver-Lake, L.; Taitt, C.; Ligler, F. Rapid detection of foodborne contaminants using an Array Biosensor. Sens. Actuators B Chem. 2006, 113, 599–607. [Google Scholar] [CrossRef]
- Turner, A. Biosensors—Sense and Sensitivity. Science 2000, 290, 1315–1317. [Google Scholar] [CrossRef] [PubMed]
- Kloth, K.; Rye-Johnsen, M.; Didier, A.; Dietrich, R.; Märtlbauer, E.; Niessner, R.; Seidel, M. A regenerable immunochip for the rapid determination of 13 different antibiotics in raw milk. Analyst 2009, 134, 1433. [Google Scholar] [CrossRef] [PubMed]
- Nicewarner-Pena, S.; Freeman, R.; Reiss, B.; He, L.; Pena, D.; Walton, J.; Cromer, R.; Keating, C.; Natan, M. Submicrometer Metallic Barcodes. Science 2001, 294, 137–141. [Google Scholar] [CrossRef] [PubMed]
- Derwinska, K.; Gheber, L.; Sauer, U.; Schorn, L.; Preininger, C. Effect of Surface Parameters on the Performance of IgG-Arrayed Hydrogel Chips: A Comprehensive Study. Langmuir 2007, 23, 10551–10558. [Google Scholar] [CrossRef] [PubMed]
- Derwinska, K.; Sauer, U.; Preininger, C. Reproducibility of hydrogel slides in on-chip immunoassays with respect to scanning mode, spot circularity, and data filtering. Anal. Biochem. 2007, 370, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Guschin, D.; Yershov, G.; Zaslavsky, A.; Gemmell, A.; Shick, V.; Proudnikov, D.; Arenkov, P.; Mirzabekov, A. Manual Manufacturing of Oligonucleotide, DNA, and Protein Microchips. Anal. Biochem. 1997, 250, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Stillman, B.; Tonkinson, J. FAST slides: A novel surface for microarrays. Biotechniques 2000, 29, 630–635. [Google Scholar] [PubMed]
- Jeronimo, P.; Araujo, A.; Montenegro, M. Optical sensors and biosensors based on sol-gel films. Talanta 2007, 72, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Zhou, X.; Hobley, J.; Su, X. Comparative Study of Random and Oriented Antibody Immobilization as Measured by Dual Polarization Interferometry and Surface Plasmon Resonance Spectroscopy. Langmuir 2012, 28, 997–1004. [Google Scholar] [CrossRef] [PubMed]
- Fowler, J.; Stuart, M.; Wong, D. Self-Assembled Layer of Thiolated Protein G as an Immunosensor Scaffold. Anal. Chem. 2007, 79, 350–354. [Google Scholar] [CrossRef] [PubMed]
- Soler, M.; Estevez, M.; Alvarez, M.; Otte, M.; Sepulveda, B.; Lechuga, L. Direct Detection of Protein Biomarkers in Human Fluids Using Site-Specific Antibody Immobilization Strategies. Sensors 2014, 14, 2239–2258. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Lee, E.; Cho, Y.; Matsui, T.; Kang, I.; Kim, T.; Han, M. ProteoChip: A highly sensitive protein microarray prepared by a novel method of protein immobilization for application of protein-protein interaction studies. Proteomics 2003, 3, 2289–2304. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, H.; Kanazaki, K.; Nakanishi, T.; Shiotsuka, H.; Hatakeyama, S.; Kaieda, M.; Imamura, T.; Umetsu, M.; Kumagai, I. Biomimetic Engineering of Modular Bispecific Antibodies for Biomolecule Immobilization. Langmuir 2011, 27, 9656–9661. [Google Scholar] [CrossRef] [PubMed]
- Poller, A.; Crooks, S.; Preininger, C. Influence of different surface chemistries on the ultrasensitive on-chip detection of enrofloxacin in milk. Sens. Actuators B Chem. 2015, 209, 1077–1083. [Google Scholar] [CrossRef]
- Sauer, U.; Domnanich, P.; Preininger, C. Protein chip for the parallel quantification of high and low abundant biomarkers for sepsis. Anal. Biochem. 2011, 419, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Soler, M.; Mesa-Antunez, P.; Estevez, M.; Ruiz-Sanchez, A.; Otte, M.; Sepulveda, B.; Collado, D.; Mayorga, C.; Torres, M.; Perez-Inestrosa, E.; et al. Highly sensitive dendrimer-based nanoplasmonic biosensor for drug allergy diagnosis. Biosens. Bioelectron. 2015, 66, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Souto, D.; Fonseca, A.; Barragan, J.; Luz, R.; Andrade, H.; Damos, F.; Kubota, L. SPR analysis of the interaction between a recombinant protein of unknown function in Leishmania infantum immobilised on dendrimers and antibodies of the visceral leishmaniasis: A potential use in immunodiagnosis. Biosens. Bioelectron. 2015, 70, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Barbey, R.; Kauffmann, E.; Ehrat, M.; Klok, H. Protein Microarrays Based on Polymer Brushes Prepared via Surface-Initiated Atom Transfer Radical Polymerization. Biomacromolecules 2010, 11, 3467–3479. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Guo, C.; Hu, W.; Lu, Z.; Li, C. Sensitive protein microarray synergistically amplified by polymer brush-enhanced immobilizations of both probe and reporter. J. Colloid Interface Sci. 2011, 360, 593–599. [Google Scholar] [CrossRef] [PubMed]
- Gheber, L.A.; Ben Gurion University, Beer Sheva, Israel. Personal communication, 2011.
- Dostalek, J.; Austrian Institute of Technology, Vienna, Austria. Personal communication, 2015.
- Cras, J.; Rowe-Taitt, C.; Nivens, D.; Ligler, F. Comparison of chemical cleaning methods of glass in preparation for silanization. Biosen. Bioelectron. 1999, 14, 683–688. [Google Scholar] [CrossRef]
- Jo, S.; Park, K. Surface modification using silanated poly(ethylene glycol)s. Biomaterials 2000, 21, 605–616. [Google Scholar] [CrossRef]
- Pallandre, A.; Glinel, K.; Jonas, A.; Nysten, B. Binary Nanopatterned Surfaces Prepared from Silane Monolayers. Nano Lett. 2004, 4, 365–371. [Google Scholar] [CrossRef]
- Köhler, G.; Milstein, C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975, 256, 495–497. [Google Scholar] [CrossRef] [PubMed]
- Hoogenboom, H.R. Selecting and screening recombinant antibody libraries. Nat. Biotechnol. 2005, 23, 1105–1116. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Shen, Z.; Mernaugh, R. Recombinant antibodies and their use in biosensors. Anal. Bioanal. Chem. 2012, 402, 3027–3038. [Google Scholar] [CrossRef] [PubMed]
- Egelhofer, T.A.; Minoda, A.; Klugman, S.; Lee, K.; Kolasinska-Zwierz, P.; Alekseyenko, A.A.; Cheung, M.S.; Day, D.S.; Gadel, S.; Gorchakov, A.A.; et al. An assessment of histone-modification antibody quality. Nat. Struct. Mol. Biol. 2011, 18, 91–93. [Google Scholar] [CrossRef] [PubMed]
- Michel, M.C.; Wieland, T.; Tsujimoto, G. How reliable are G-protein-coupled receptor antibodies? NaunynSchmiedeberg's Arch. Pharmacol. 2009, 379, 385–388. [Google Scholar] [CrossRef] [PubMed]
- Peltomaa, R.; Barderas, R.; Sauer, U.; Benito-Peña, E.; Moreno-Bondi, M.C. Microarray-based immunoassay with synthetic mimotopes for the detection of fumonisin. Biosens. Bioelectron. 2017, in press. [Google Scholar]
- Liu, Q.; Wang, J.; Boyd, B. Peptide-based biosensors. Talanta 2015, 136, 114–127. [Google Scholar] [CrossRef] [PubMed]
- Mairal, T.; Cengiz Özalp, V.; Lozano Sánchez, P.; Mir, M.; Katakis, I.; O’Sullivan, C. Aptamers: Molecular tools for analytical applications. Anal. Bioanal. Chem. 2007, 390, 989–1007. [Google Scholar] [CrossRef] [PubMed]
- Lakhin, A.V.; Tarantul, V.Z.; Gening, L.V. Aptamers: Problems, Solutions and Prospects. Acta Nat. 2013, 5, 19–34. [Google Scholar]
- Noxxon Pharma: Spiegelmers. Available online: http://www.noxxon.com/index.php?option=com_content&view=article&id=13&Itemid=529 (accessed on 25 November 2016).
- Pultar, J.; Sauer, U.; Domnanich, P.; Preininger, C. Aptamer-antibody on-chip sandwich immunoassay for detection of CRP in spiked serum. Biosens. Bioelectron. 2009, 24, 1456–1461. [Google Scholar] [CrossRef] [PubMed]
- Uzun, L.; Turner, A. Molecularly-imprinted polymer sensors: Realising their potential. Biosens. Bioelectron. 2016, 76, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Haupt, K.; Mosbach, K. Plastic antibodies: Developments and applications. Trends Biotechnol. 1998, 16, 468–475. [Google Scholar] [CrossRef]
- Haupt, K.; Mosbach, K. Molecularly Imprinted Polymers and Their Use in Biomimetic Sensors. Chem. Rev. 2000, 100, 2495–2504. [Google Scholar] [CrossRef] [PubMed]
- Piletsky, S.A.; Turner, N.W.; Laitenberger, P. Molecularly imprinted polymers in clinical diagnostics—Future potential and existing problems. Med. Eng. Phys. 2006, 28, 971–977. [Google Scholar] [CrossRef] [PubMed]
- Dickert, F.; Lieberzeit, P.; Hayden, O. Sensor strategies for microorganism detection—from physical principles to imprinting procedures. Anal. Bioanal. Chem. 2003, 377, 540–549. [Google Scholar] [CrossRef] [PubMed]
- Cohen, T.; Starosvetsky, J.; Cheruti, U.; Armon, R. Whole Cell Imprinting in Sol-Gel Thin Films for Bacterial Recognition in Liquids: Macromolecular Fingerprinting. Int. J. Mol. Sci. 2010, 11, 1236–1252. [Google Scholar] [CrossRef] [PubMed]
- Bolisay, L.; Culver, J.; Kofinas, P. Molecularly imprinted polymers for tobacco mosaic virus recognition. Biomaterials 2006, 27, 4165–4168. [Google Scholar] [CrossRef] [PubMed]
- Buchegger, P.; Lieberzeit, P.; Preininger, C. Thermo-Nanoimprinted Biomimetic Probe for LPS and LTA Immunosensing. Anal. Chem. 2014, 86, 1679–1686. [Google Scholar] [CrossRef] [PubMed]
- McWilliam, I.; Chong Kwan, M.; Hall, D. Inkjet Printing for the Production of Protein Microarrays. In Protein Microarrays: Methods and Protocols, 1st ed.; Korf, U., Ed.; Humana Press: New York, NY, USA, 2011; pp. 345–361. [Google Scholar]
- Odom, T.; Love, J.; Wolfe, D.; Paul, K.; Whitesides, G. Improved Pattern Transfer in Soft Lithography Using Composite Stamps. Langmuir 2002, 18, 5314–5320. [Google Scholar] [CrossRef]
- Alom Ruiz, S.; Chen, C. Microcontact printing: A tool to pattern. Soft Matter 2007, 3, 168–177. [Google Scholar] [CrossRef]
- Tan, J.; Tien, J.; Chen, C. Microcontact Printing of Proteins on Mixed Self-Assembled Monolayers. Langmuir 2002, 18, 519–523. [Google Scholar] [CrossRef]
- Lu, H.; Homola, J.; Campbell, C.; Nenninger, G.; Yee, S.; Ratner, B. Protein contact printing for a surface plasmon resonance biosensor with on-chip referencing. Sens. Actuators B Chem. 2001, 74, 91–99. [Google Scholar] [CrossRef]
- Cau, J.; Ludovic, L.; Marie, N.; Adriana, L.; Vincent, P. Magnetic field assisted microcontact printing: A new concept of fully automated and calibrated process. Microelectron. Eng. 2013, 110, 207–214. [Google Scholar] [CrossRef]
- Taha, H.; Marks, R.; Gheber, L.; Rousso, I.; Newman, J.; Sukenik, C.; Lewis, A. Protein printing with an atomic force sensing nanofountainpen. Appl. Phys. Lett. 2003, 83, 1041. [Google Scholar] [CrossRef]
- Gheber, L. Nano Fountain Pen: Toward Integrated, Portable, Lab-on-Chip Devices. In Biological Applications of Microfluidics, 1st ed.; Gomez, F., Ed.; John Wiley Sons Inc.: New York, NY, USA, 2008; pp. 369–384. [Google Scholar]
- Lee, K.; Kim, E.; Mirkin, C.; Wolinsky, S. The Use of Nanoarrays for Highly Sensitive and Selective Detection of Human Immunodeficiency Virus Type 1 in Plasma. Nano Lett. 2004, 4, 1869–1872. [Google Scholar] [CrossRef]
- Ellmark, P.; Ghatnekar-Nilsson, S.; Meister, A.; Heinzelmann, H.; Montelius, L.; Wingren, C.; Borrebaeck, C. Attovial-based antibody nanoarrays. Proteomics 2009, 9, 5406–5413. [Google Scholar] [CrossRef] [PubMed]
- Petersson, L.; Berthet Duroure, N.; Auger, A.; Dexlin-Mellby, L.; Borrebaeck, C.; Ait Ikhlef, A.; Wingren, C. Generation of miniaturized planar ecombinant antibody arrays using a microcantilever-based printer. Nanotechnology 2014, 25, 275104. [Google Scholar] [CrossRef] [PubMed]
- Sauer, U.; Pultar, J.; Preininger, C. Critical role of the sample matrix in a point-of-care protein chip for sepsis. J. Immunol. Methods 2012, 378, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Gogalic, S.; Sauer, U.; Doppler, S.; Preininger, C. Bladder cancer biomarker array to detect aberrant levels of proteins in urine. Analyst 2015, 140, 724–735. [Google Scholar] [CrossRef] [PubMed]
- Kunze, A.; Pei, L.; Elsässer, D.; Niessner, R.; Seidel, M. High performance concentration method for viruses in drinking water. J. Virol. Methods 2015, 222, 132–137. [Google Scholar] [CrossRef] [PubMed]
- Carion, O.; Souplet, V.; Olivier, C.; Maillet, C.; Medard, N.; El-Mahdi, O.; Durand, J.; Melnyk, O. Chemical Micropatterning of Polycarbonate for Site-Specific Peptide Immobilization and Biomolecular Interactions. ChemBioChem 2007, 8, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Corgier, B.P.; Mandon, C.A.; LeGoff, G.C.; Blum, L.J.; Marquette, C.A. Adhesive microarrays for multipurpose diagnostic tools. Lab Chip 2011, 11, 3006–3010. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Bai, Y.; Ge, Q.; Zhou, S.; Wen, T.; Lu, Z. Microarray-in-a-Tube for Detection of Multiple Viruses. Clin. Chem. 2006, 53, 188–194. [Google Scholar] [CrossRef] [PubMed]
- Baldini, F.; Bolzoni, L.; Giannetti, A.; Kess, M.; Krämer, P.; Kremmer, E.; Porro, G.; Senesi, F.; Trono, C. A new procalcitonin optical immunosensor for POCT applications. Anal. Bioanal. Chem. 2009, 393, 1183–1190. [Google Scholar] [CrossRef] [PubMed]
- Buchegger, P.; Sauer, U.; Toth-Székély, H.; Preininger, C. Miniaturized Protein Microarray with Internal Calibration as Point-of-Care Device for Diagnosis of Neonatal Sepsis. Sensors 2012, 12, 1494–1508. [Google Scholar] [CrossRef] [PubMed]
- Sano, T.; Smith, C.L.; Cantor, C.R. Immuno-PCR: Very sensitive antigen detection by means of specific antibody-DNA conjugates. Science 1992, 258, 120–122. [Google Scholar] [CrossRef]
- Schweitzer, B.; Wiltshire, S.; Lambert, J.; O’Malley, S.; Kukanskis, K.; Zhu, Z.; Kingsmore, S.; Lizardi, P.; Ward, D. Immunoassays with rolling circle DNA amplification: A versatile platform for ultrasensitive antigen detection. PNAS 2000, 97, 10113–10119. [Google Scholar] [CrossRef] [PubMed]
- Vikesland, P.; Wigginton, K. Nanomaterial Enabled Biosensors for Pathogen Monitoring—A Review. Environ. Sci. Technol. 2010, 44, 3656–3669. [Google Scholar] [CrossRef] [PubMed]
- Tschmelak, J.; Proll, G.; Gauglitz, G. Verification of performance with the automated direct optical TIRF immunosensor (River Analyser) in single and multi-analyte assays with real water samples. Biosens. Bioelectron. 2004, 20, 743–752. [Google Scholar] [CrossRef] [PubMed]
- Nagl, S.; Bauer, R.; Sauer, U.; Preininger, C.; Bogner, U.; Schaeferling, M. Microarray analysis of protein–protein interactions based on FRET using subnanosecond-resolved fluorescence lifetime imaging. Biosens. Bioelectron. 2008, 24, 397–402. [Google Scholar] [CrossRef] [PubMed]
- Wen, J.; Shi, X.; He, Y.; Zhou, J.; Li, Y. Novel plastic biochips for colorimetric detection of biomolecules. Anal. Bioanal. Chem. 2012, 404, 1935–1944. [Google Scholar] [CrossRef] [PubMed]
- Weller, M.; Schuetz, A.; Winklmair, M.; Niessner, R. Highly parallel affinity sensor for the detection of environmental contaminants in water. Anal. Chim. Acta 1999, 393, 29–41. [Google Scholar] [CrossRef]
- Homola, J.; Vaisocherová, H.; Dostálek, J.; Piliarik, M. Multi-analyte surface plasmon resonance biosensing. Methods 2005, 37, 26–36. [Google Scholar] [CrossRef] [PubMed]
- Mirasoli, M.; Bonvicini, F.; Dolci, L.; Zangheri, M.; Gallinella, G.; Roda, A. Portable chemiluminescence multiplex biosensor for quantitative detection of three B19 DNA genotypes. Anal. Bioanal. Chem. 2012, 405, 1139–1143. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Hagen, J.; Papautsky, I. Point-of-care colorimetric detection with a smartphone. Lab Chip 2012, 12, 4240. [Google Scholar] [CrossRef] [PubMed]
- Schäferling, M.; Nagl, S. Optical technologies for the read out and quality control of DNA and protein microarrays. Anal. Bioanal. Chem. 2006, 385, 500–517. [Google Scholar] [CrossRef] [PubMed]
- Nam, J.; Thaxton, C.; Mirkin, C. Nanoparticle-Based Bio-Bar Codes for the Ultrasensitive Detection of Proteins. Science 2003, 301, 1884–1886. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Yuan, L.; Hua, X.; Xu, L.; Liu, S. Signal amplification strategies for DNA and protein detection based on polymeric nanocomposites and polymerization: A review. Anal. Chim. Acta 2015, 877, 19–32. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Yan, F.; Ji, H.; Wong, D.; Ju, H. Quantum-Dot-Functionalized Poly(styrene-co-acrylic acid) Microbeads: Step-Wise Self-Assembly, Characterization, and Applications for Sub-femtomolar Electrochemical Detection of DNA Hybridization. Adv. Funct. Mater. 2010, 20, 1173–1179. [Google Scholar] [CrossRef]
- Resch-Genger, U.; Grabolle, M.; Cavaliere-Jaricot, S.; Nitschke, R.; Nann, T. Quantum dots versus organic dyes as fluorescent labels. Nat. Methods 2008, 5, 763–775. [Google Scholar] [CrossRef] [PubMed]
- Dostalek, J. Plasmonic Amplification for Fluorescence Bioassays Utilizing Propagating Surface Plasmons. In Encyclopedia of Nanotechnology; Bharat, B., Ed.; Springer Science + Business Media: Dordrecht, The Netherlands, 2015; pp. 1–11. [Google Scholar]
- Bauch, M.; Hageneder, S.; Dostalek, J. Plasmonic amplification for bioassays with epi-fluorescence readout. Opt. Express 2014, 22, 32026–32038. [Google Scholar] [CrossRef] [PubMed]
- Wingren, C.; Borrebaeck, C. Progress in miniaturization of protein arrays-a step closer to high-density nanoarrays. Drug Discov. Today 2007, 12, 813–819. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Bilgin, M.; Bangham, R.; Hall, D.; Casamayor, A.; Bertone, P.; Lan, N.; Jansen, R.; Bidlingmaier, S.; Houfek, T.; et al. Global Analysis of Protein Activities Using Proteome Chips. Science 2001, 293, 2101–2105. [Google Scholar] [CrossRef] [PubMed]
- Situma, C.; Hashimoto, M.; Soper, S. Merging microfluidics with microarray-based bioassays. Biomol. Eng. 2006, 23, 213–231. [Google Scholar] [CrossRef] [PubMed]
- Gehring, A.; Albin, D.; Reed, S.; Tu, S.; Brewster, J. An antibody microarray, in multiwell plate format, for multiplex screening of foodborne pathogenic bacteria and biomolecules. Anal. Bioanal. Chem. 2008, 391, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Delehanty, J.; Ligler, F. A Microarray Immunoassay for Simultaneous Detection of Proteins and Bacteria. Anal. Chem. 2002, 74, 5681–5687. [Google Scholar] [CrossRef] [PubMed]
- Rowe, C.; Tender, L.; Feldstein, M.; Golden, J.; Scruggs, S.; MacCraith, B.; Cras, J.; Ligler, F. Array Biosensor for Simultaneous Identification of Bacterial, Viral, and Protein Analytes. Anal. Chem. 1999, 71, 3846–3852. [Google Scholar] [CrossRef] [PubMed]
- Shriver-Lake, L.; Taitt, C.; Ligler, F. Application of array biosensor for detection of food allergens. J. AOAC Int. 2004, 87, 1498–1502. [Google Scholar] [PubMed]
- Knecht, B.G.; Strasser, A.; Dietrich, R.; Märtlbauer, E.; Niessner, R.; Weller, M.G. Automated Microarray System for the Simultaneous Detection of Antibiotics in Milk. Anal. Chem. 2004, 76, 646–654. [Google Scholar] [CrossRef] [PubMed]
- Díaz-González, M.; Salvador, J.P.; Bonilla, D.; Marco, M.P.; Fernández-Sánchez, C.; Baldi, A. A microfluidic device for the automated electrical readout of low-density glass-slide microarrays. Biosens. Bioelectron. 2015, 74, 698–704. [Google Scholar] [CrossRef] [PubMed]
- Kemmler, M.; Koger, B.; Sulz, G.; Sauer, U.; Schleicher, E.; Preininger, C.; Brandenburg, A. Compact point-of-care system for clinical diagnostics. Sens. Actuators B Chem. 2009, 139, 44–51. [Google Scholar] [CrossRef]
- Kozma, P.; Lehmann, A.; Wunderlich, K.; Michel, D.; Schumacher, S.; Ehrentreich-Föster, E.; Bier, F.F. A novel handheld fluorescent microarray reader for point-of-care diagnostic. Biosens. Bioelectron. 2013, 47, 415–420. [Google Scholar] [CrossRef] [PubMed]
- Selma Gogalic, S.; Sauer, U.; Doppler, S.; Heinzel, A.; Perco, P.; Lukas, A.; Simpson, G.; Pandha, H.; Horvath, A.; Preininger, C. Validation of a protein panel for the noninvasive detection of recurrent non-muscle invasive bladder cancer. Biomarkers 2017. [Google Scholar] [CrossRef]
- De Paoli, M.; Gogalic, S.; Sauer, U.; Preininger, P.; Pandha, H.; Simpson, G.; Horvath, A.; Marquette, C. Multiplatform Biomarker Discovery for Bladder Cancer Recurrence Diagnosis. Dis. Markers 2016, 2016, 4591910. [Google Scholar] [CrossRef] [PubMed]



© 2017 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sauer, U. Analytical Protein Microarrays: Advancements Towards Clinical Applications. Sensors 2017, 17, 256. https://doi.org/10.3390/s17020256
Sauer U. Analytical Protein Microarrays: Advancements Towards Clinical Applications. Sensors. 2017; 17(2):256. https://doi.org/10.3390/s17020256
Chicago/Turabian StyleSauer, Ursula. 2017. "Analytical Protein Microarrays: Advancements Towards Clinical Applications" Sensors 17, no. 2: 256. https://doi.org/10.3390/s17020256
APA StyleSauer, U. (2017). Analytical Protein Microarrays: Advancements Towards Clinical Applications. Sensors, 17(2), 256. https://doi.org/10.3390/s17020256