
Fabrication of a Horizontal and a Vertical Large Surface Area Nanogap Electrochemical Sensor



Abstract
:1. Introduction
2. Fabrication of Horizontal and Vertical Coplanar Nanogap Sensors
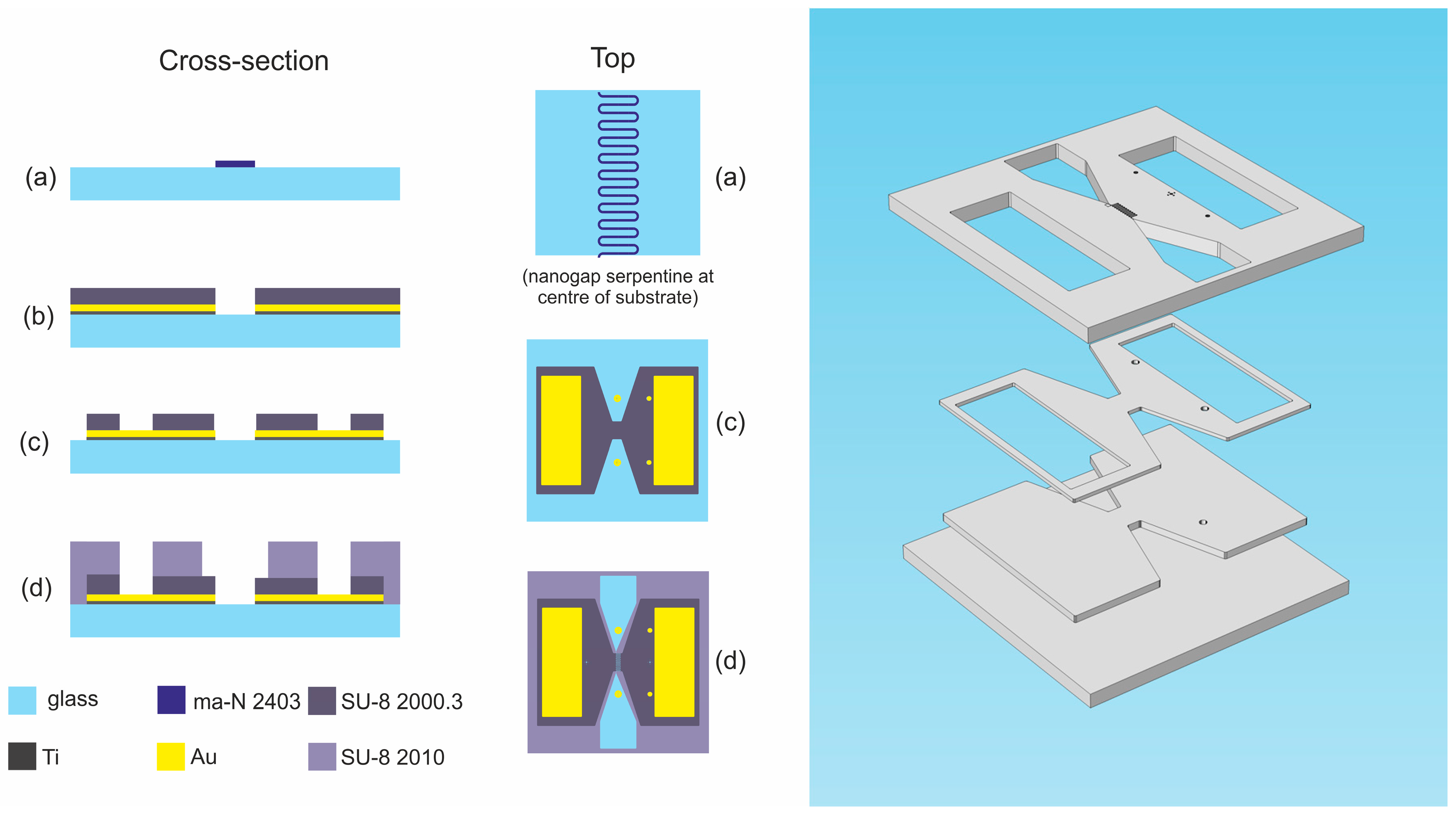
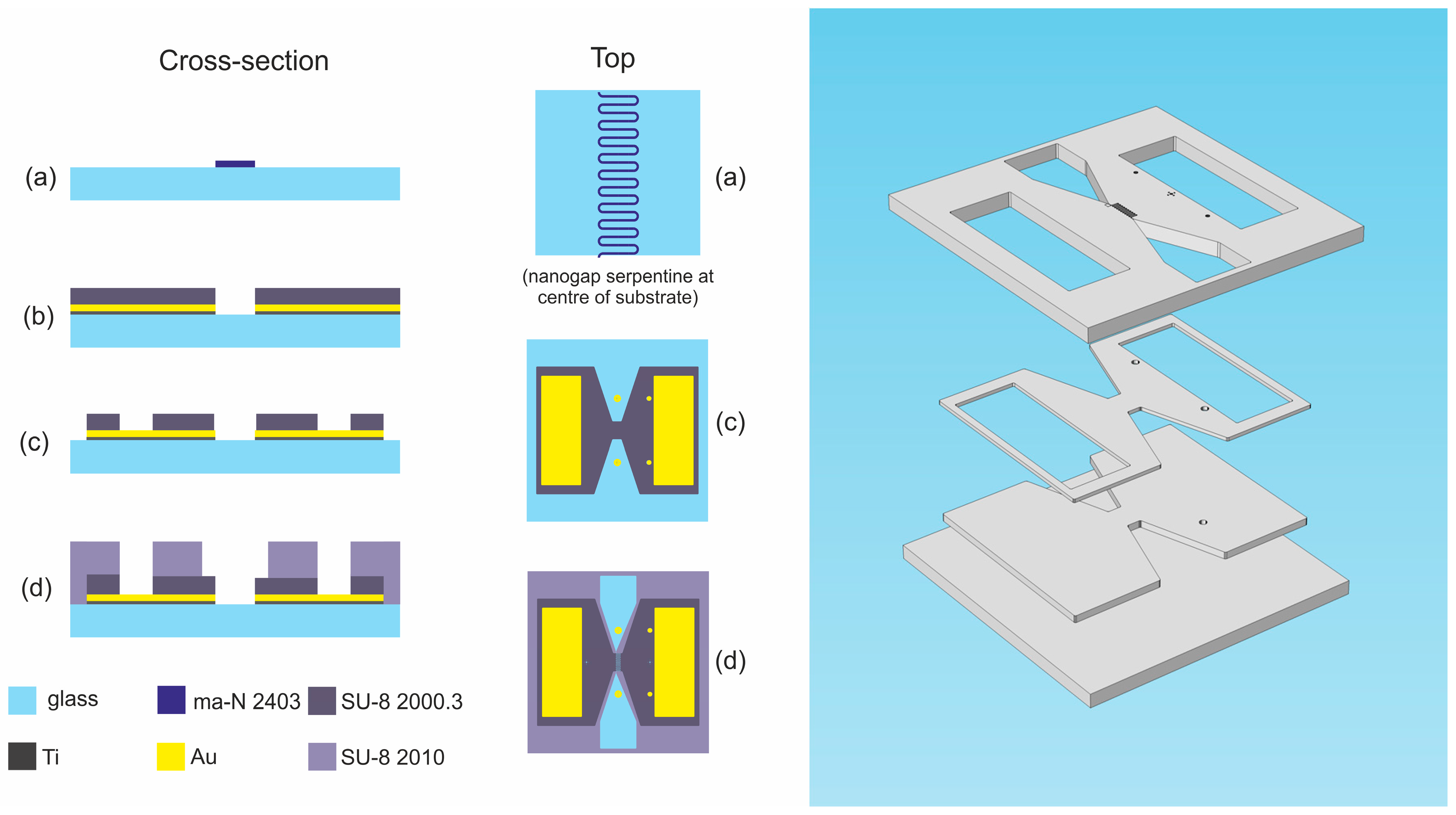
2.1. Horizontal Coplanar Nanogap Device
2.1.1. Device Details
2.1.2. Fabrication Procedure
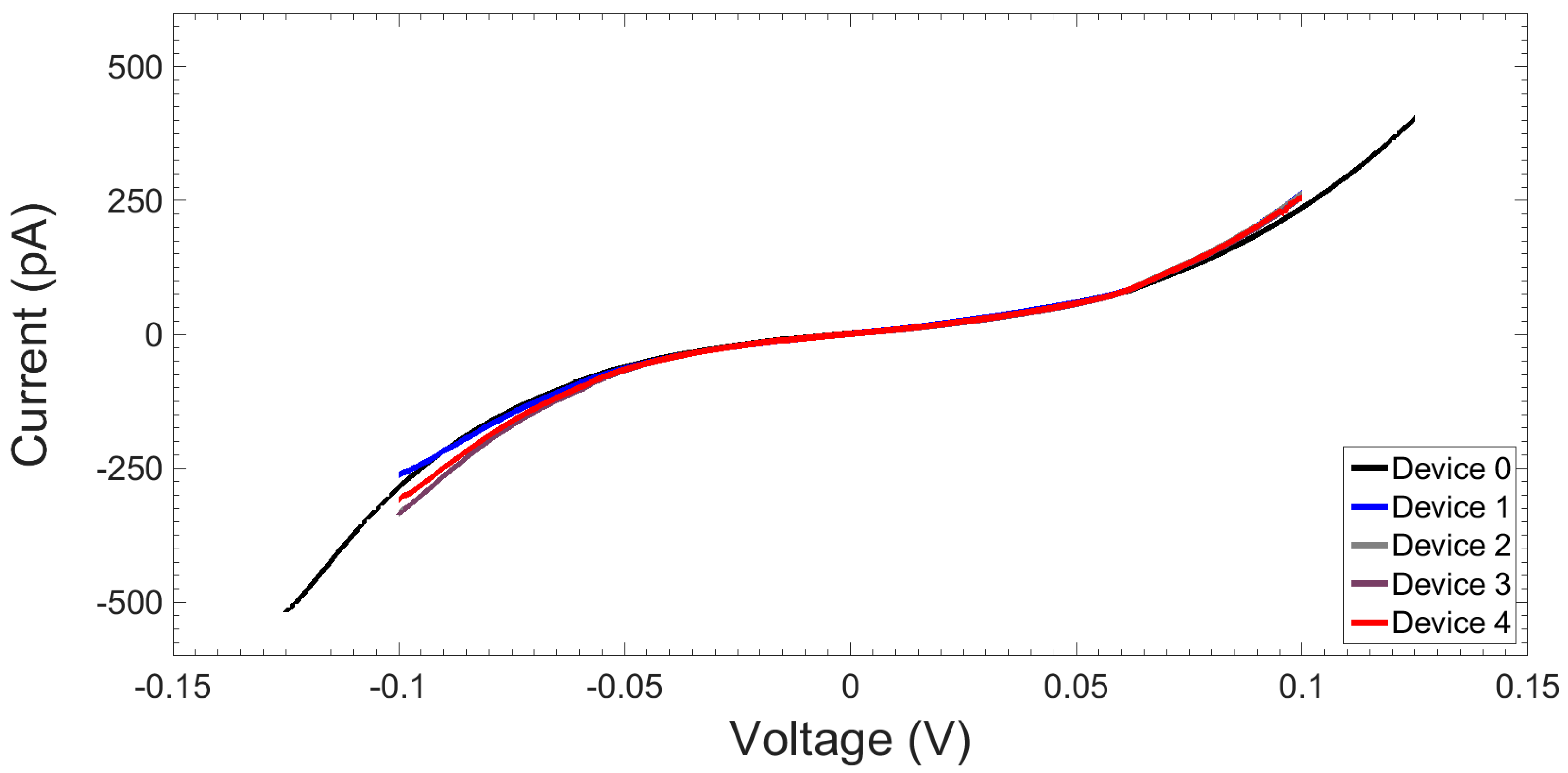
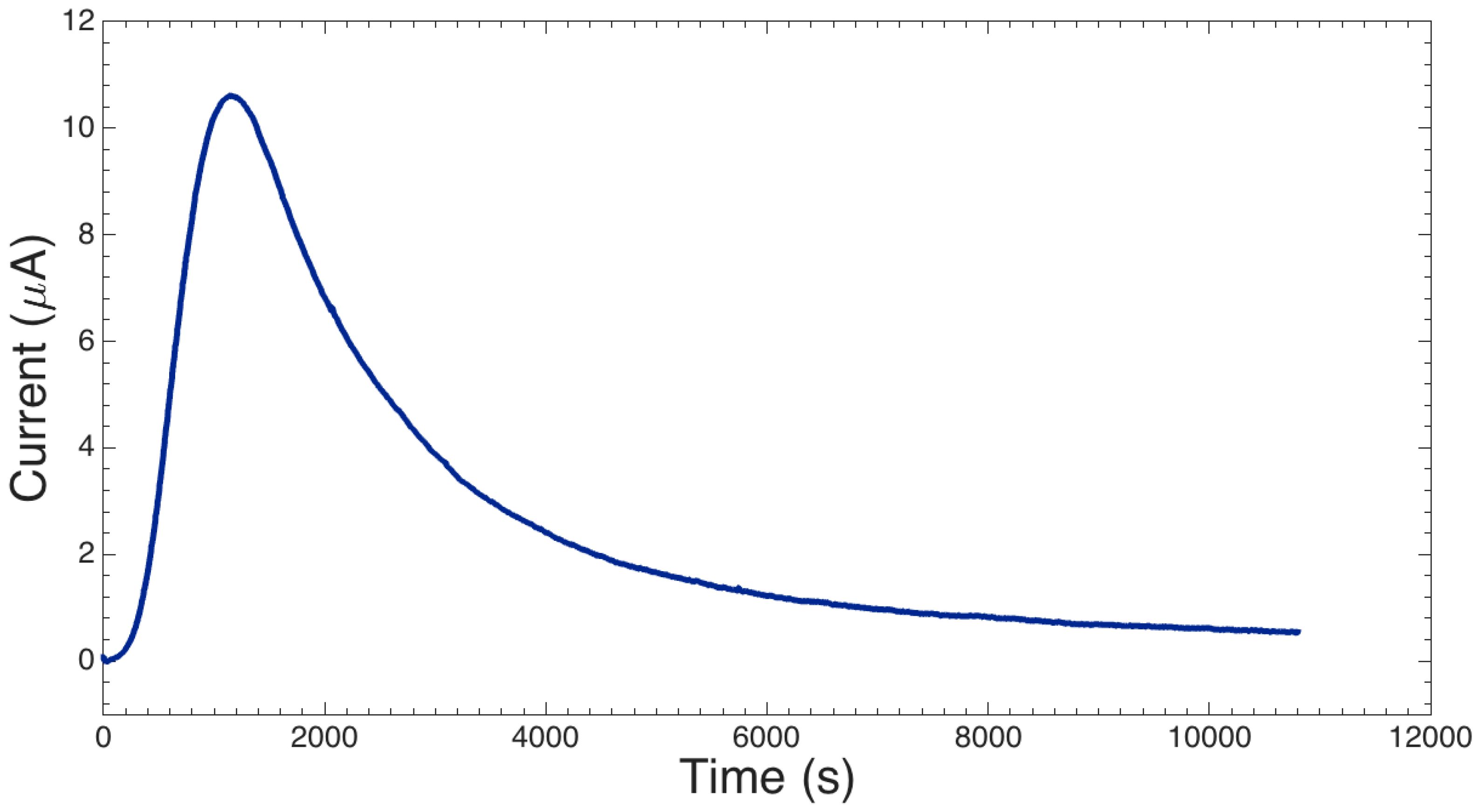
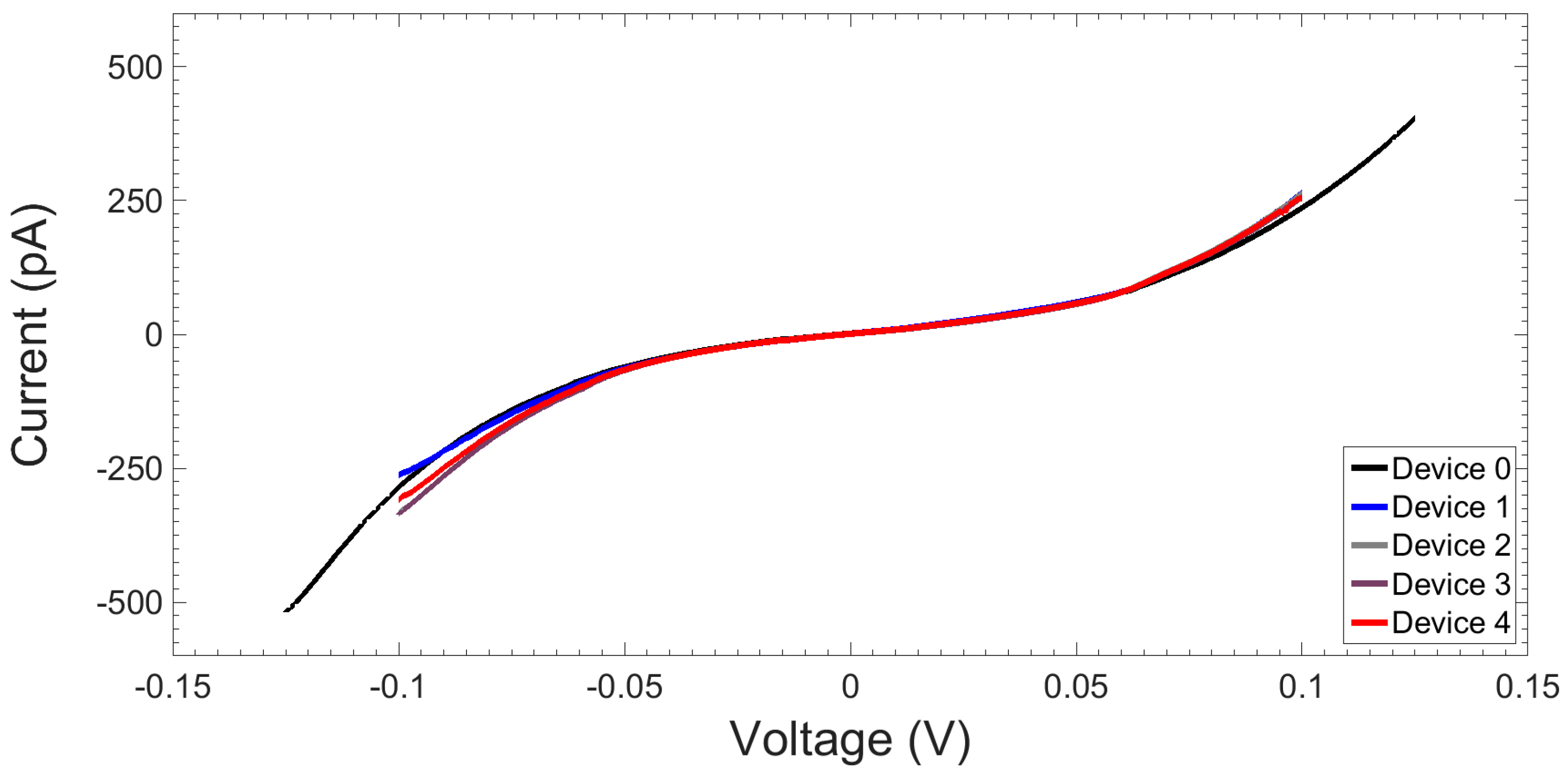
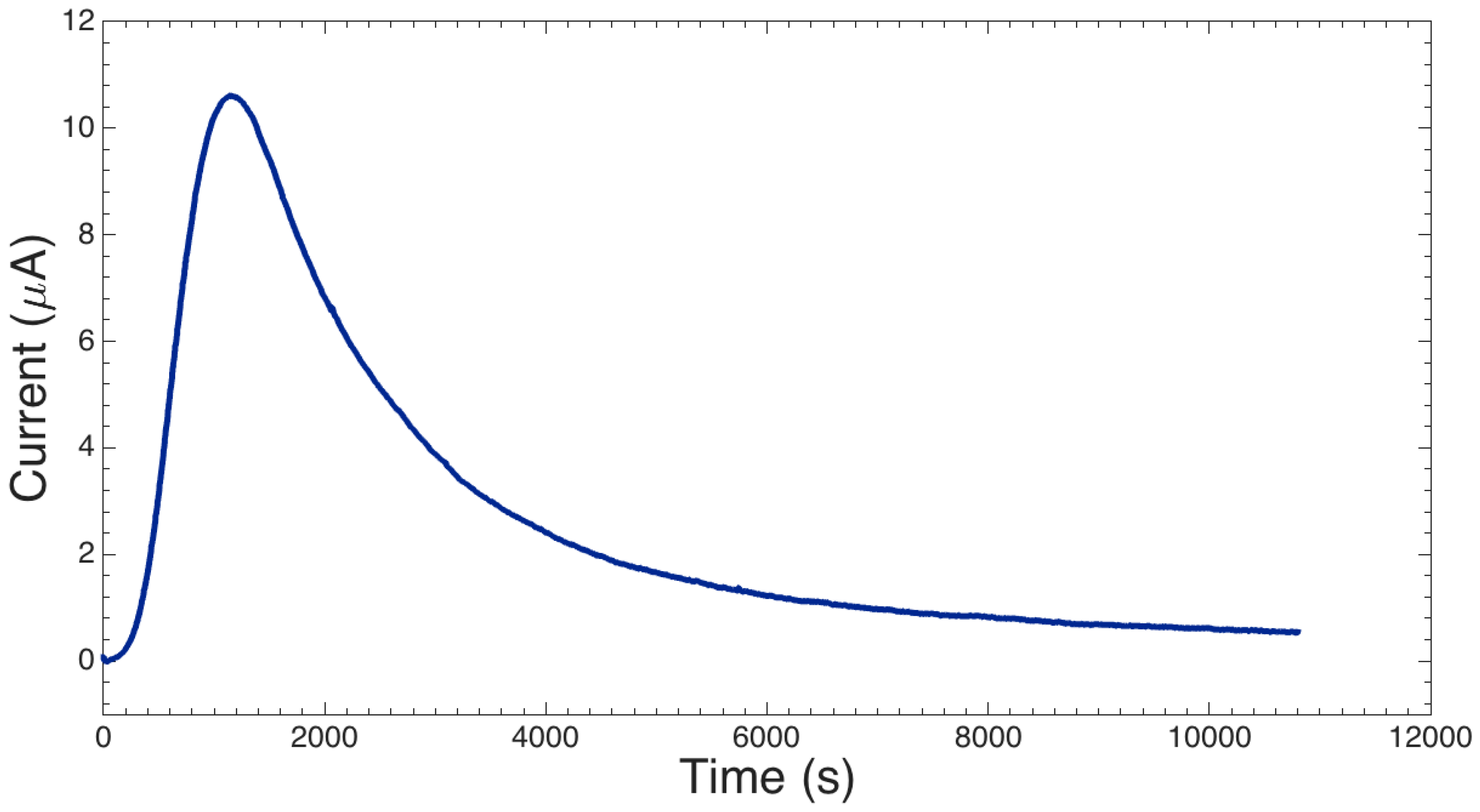
2.1.3. I-V Response
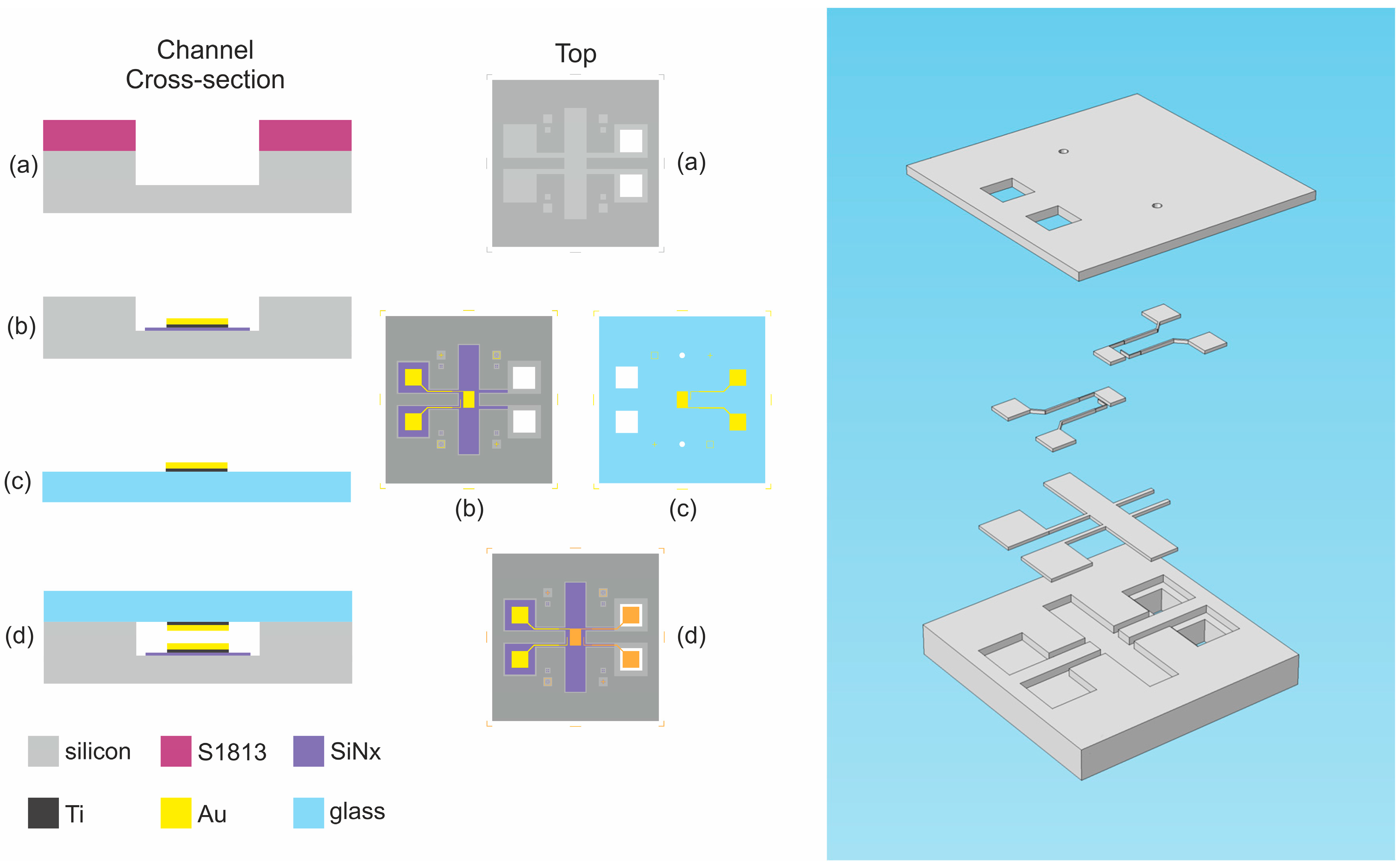
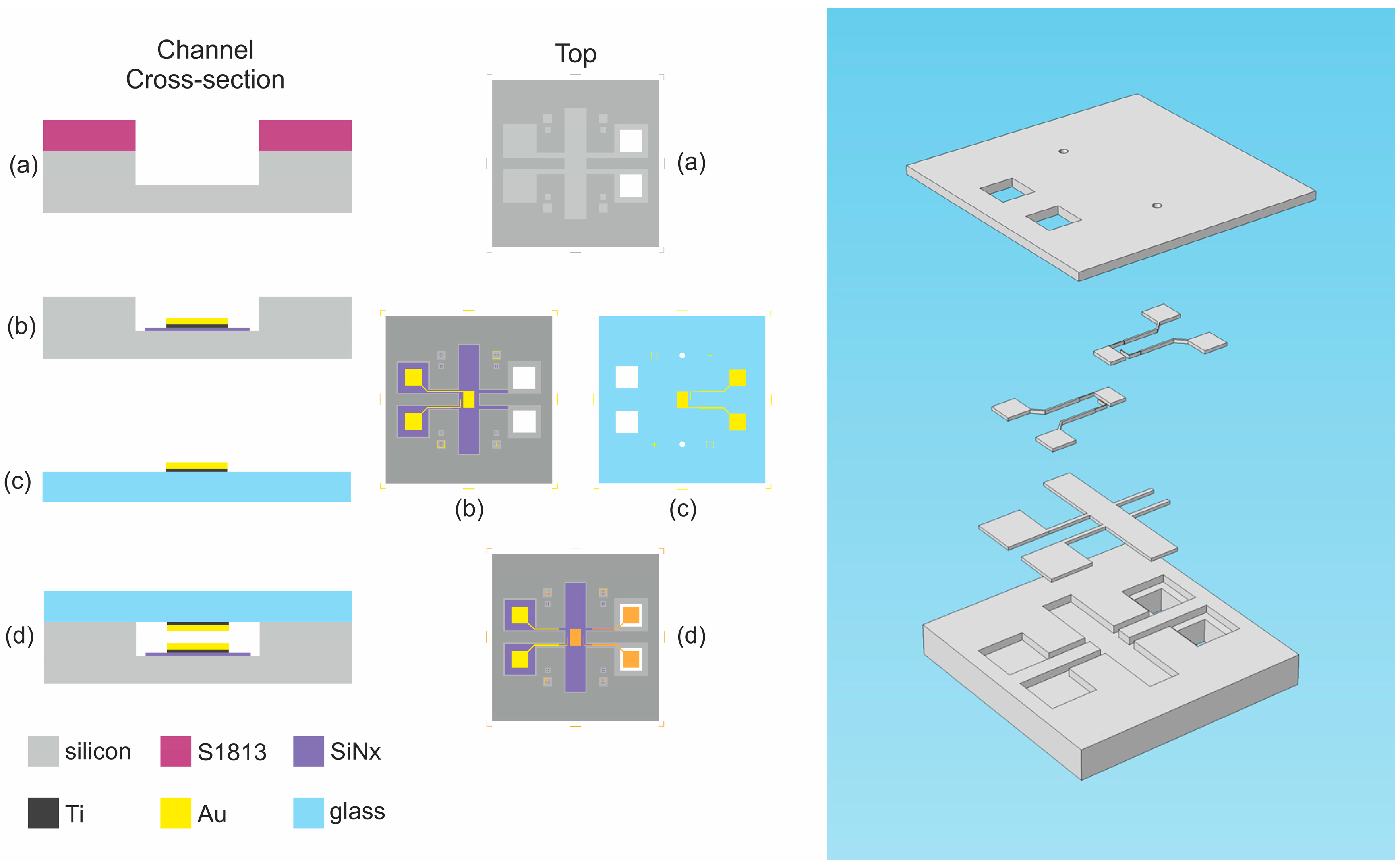
2.2. Vertical Coplanar Nanogap Device
2.2.1. Device Details
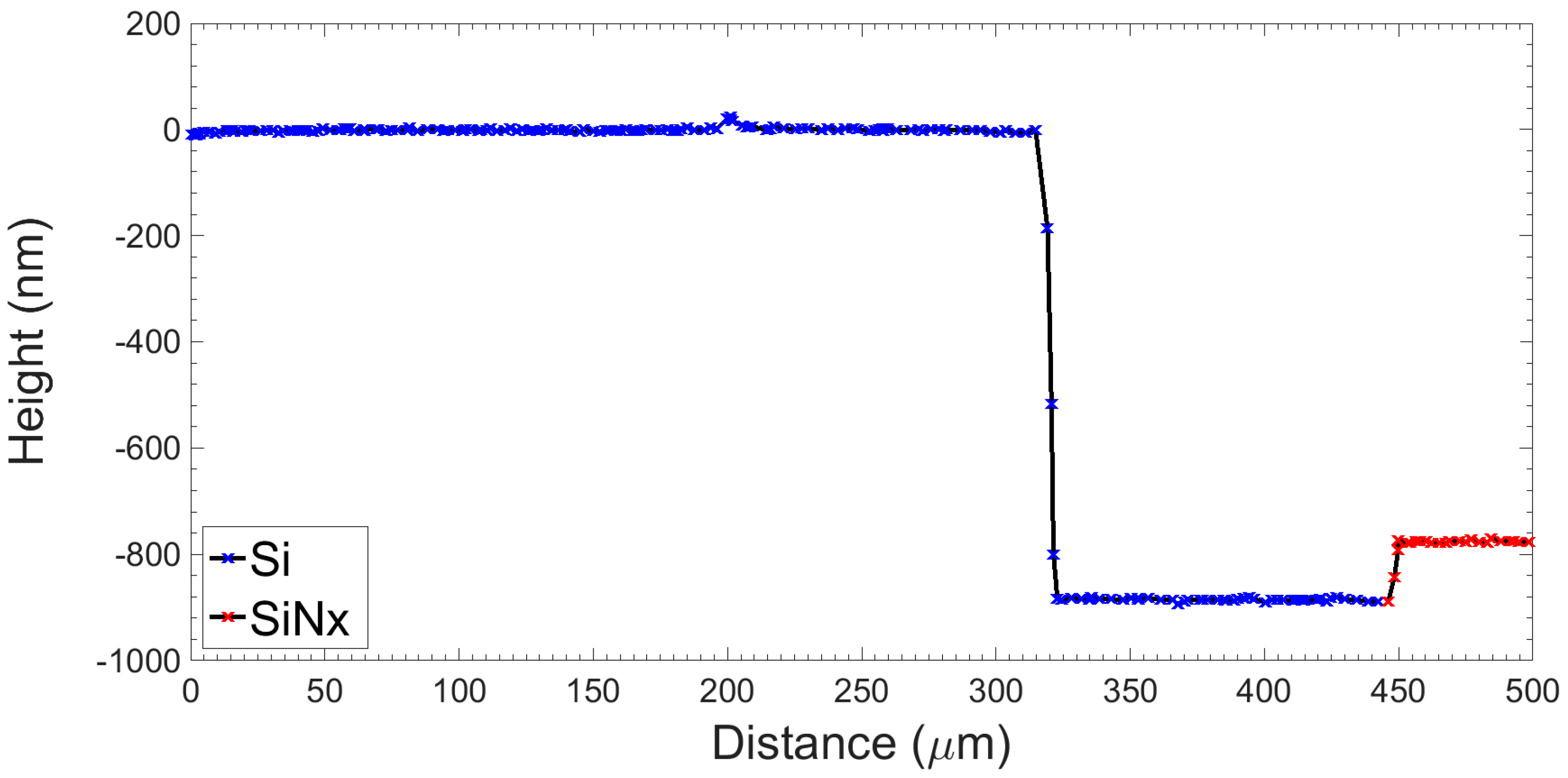
2.2.2. Fabrication Procedure
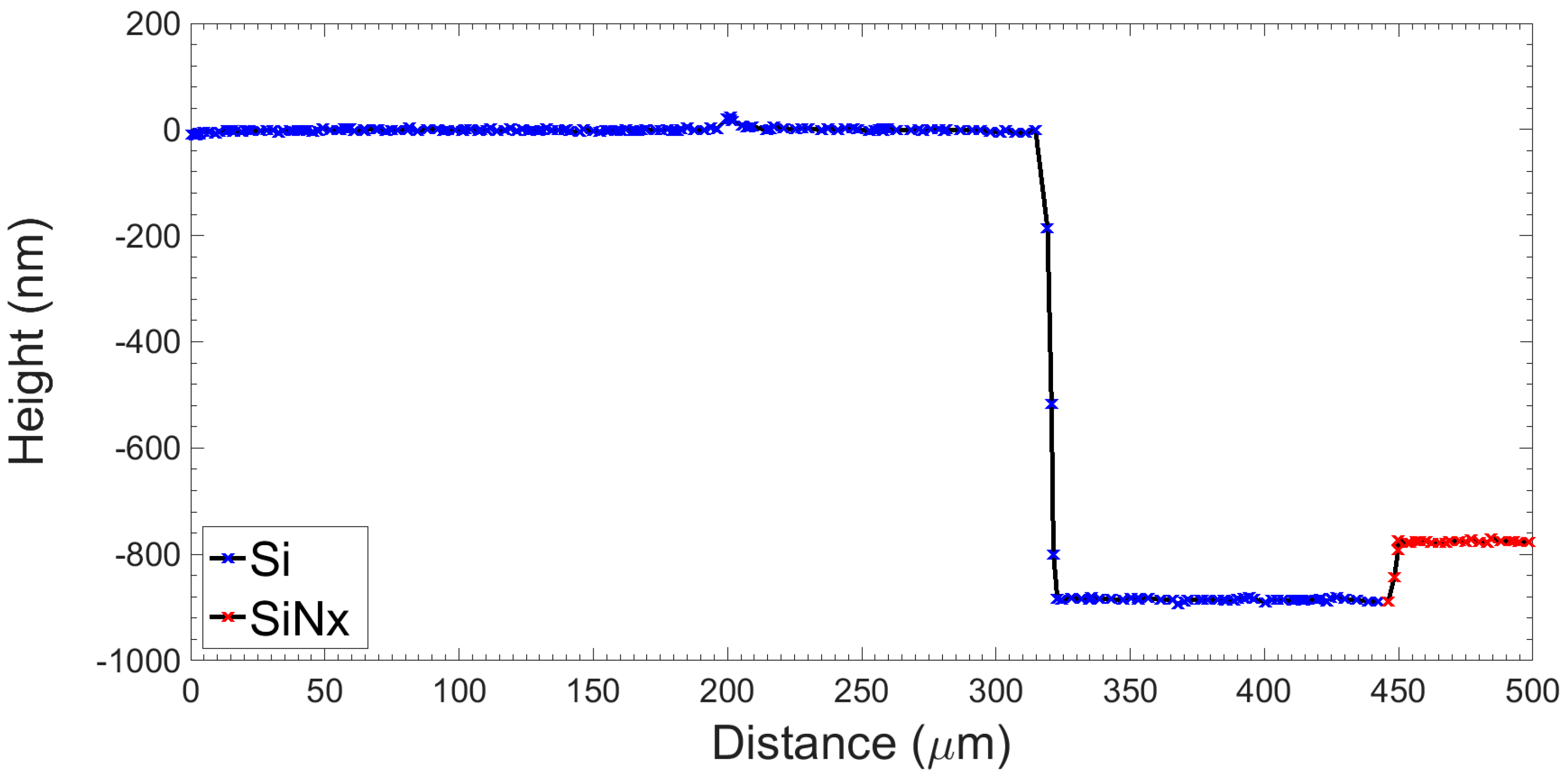
2.2.3. Anodic Bonding
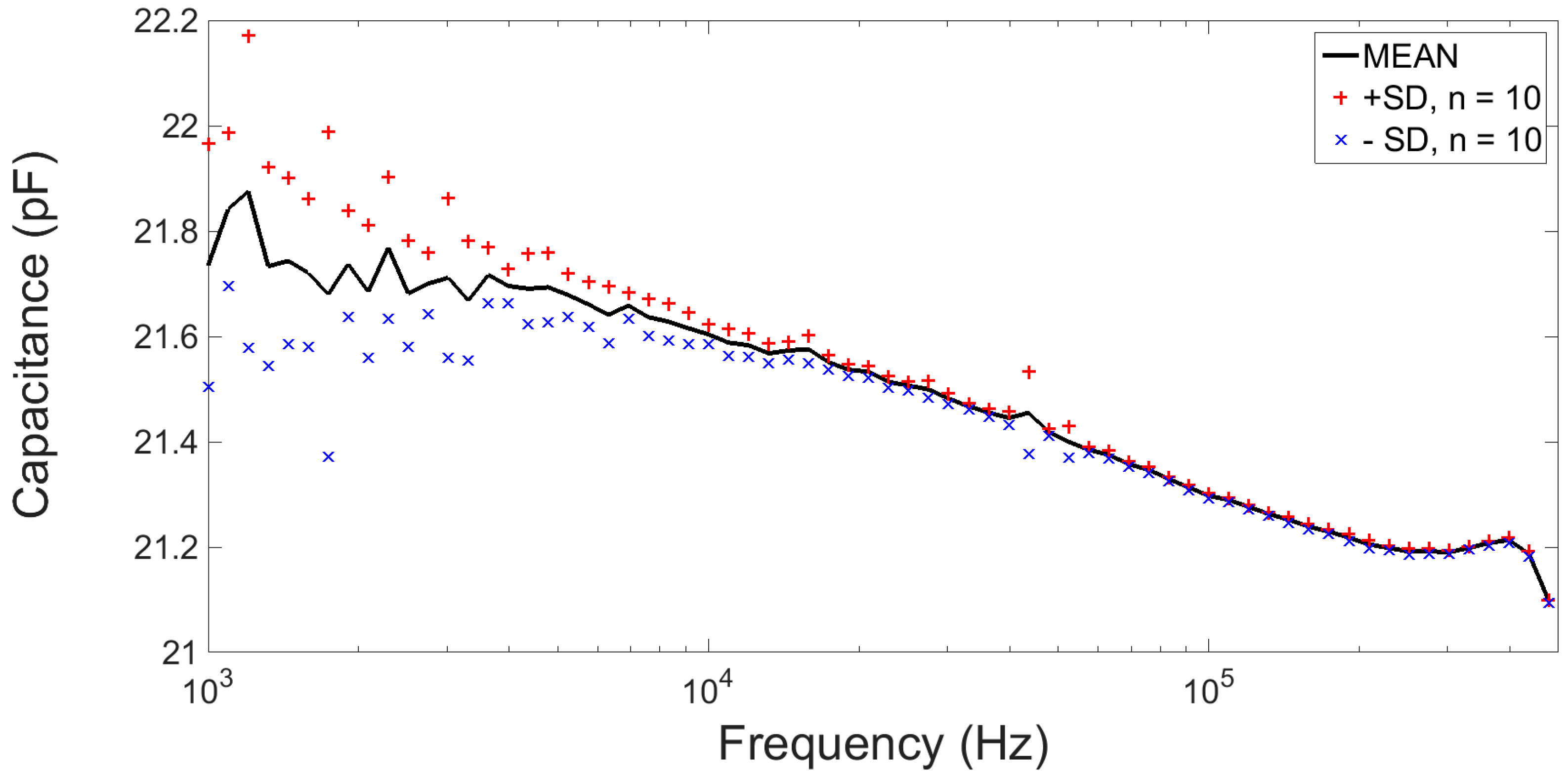
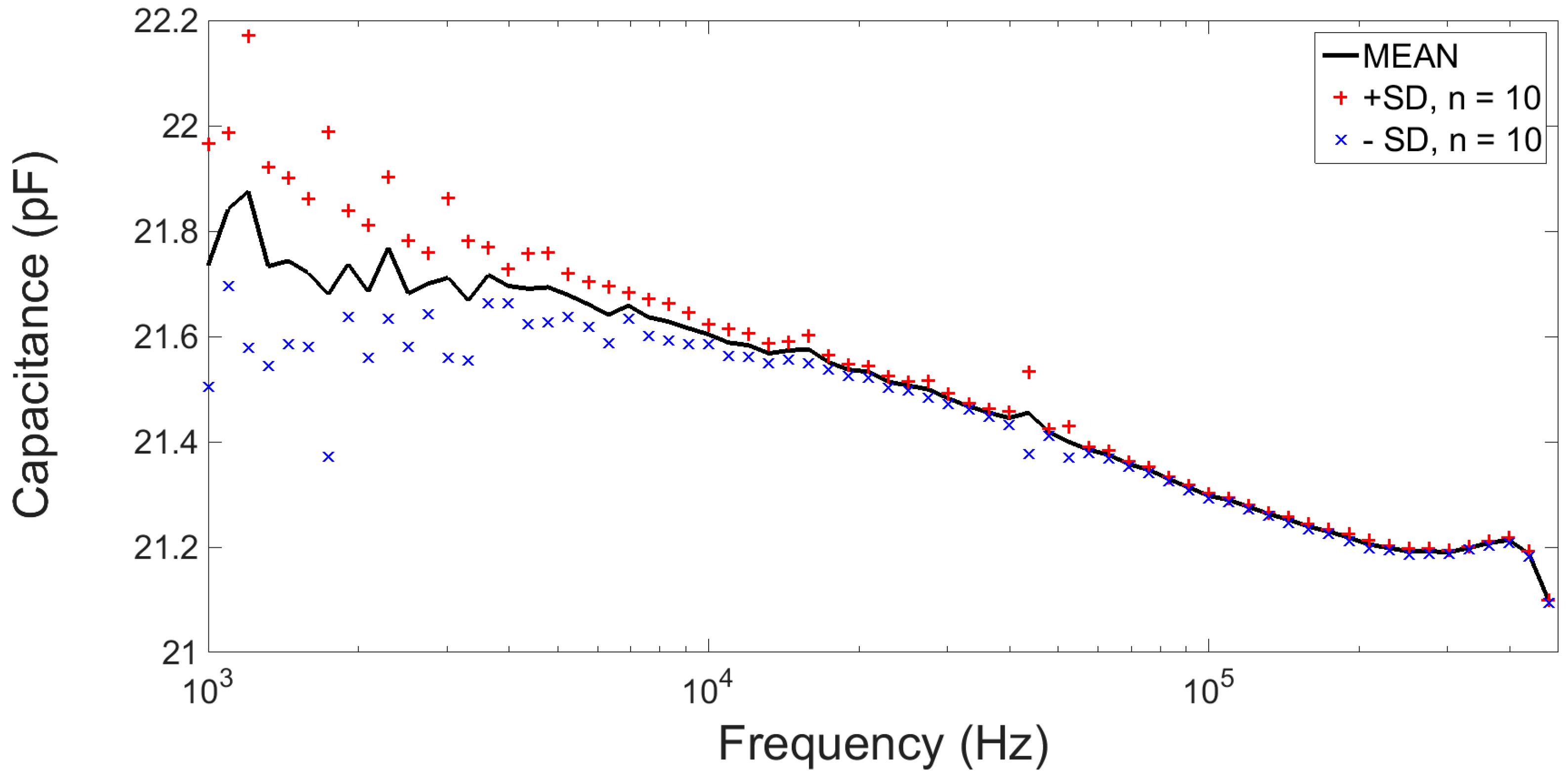
2.2.4. Electrical Characterisation
3. Dielectric Spectroscopy Sensing of DNA
3.1. Detection Scheme
3.2. Experimental Details
3.2.1. Reagents
3.2.2. Probe Layer Fabrication
3.2.3. Dielectric Spectroscopy Measurements
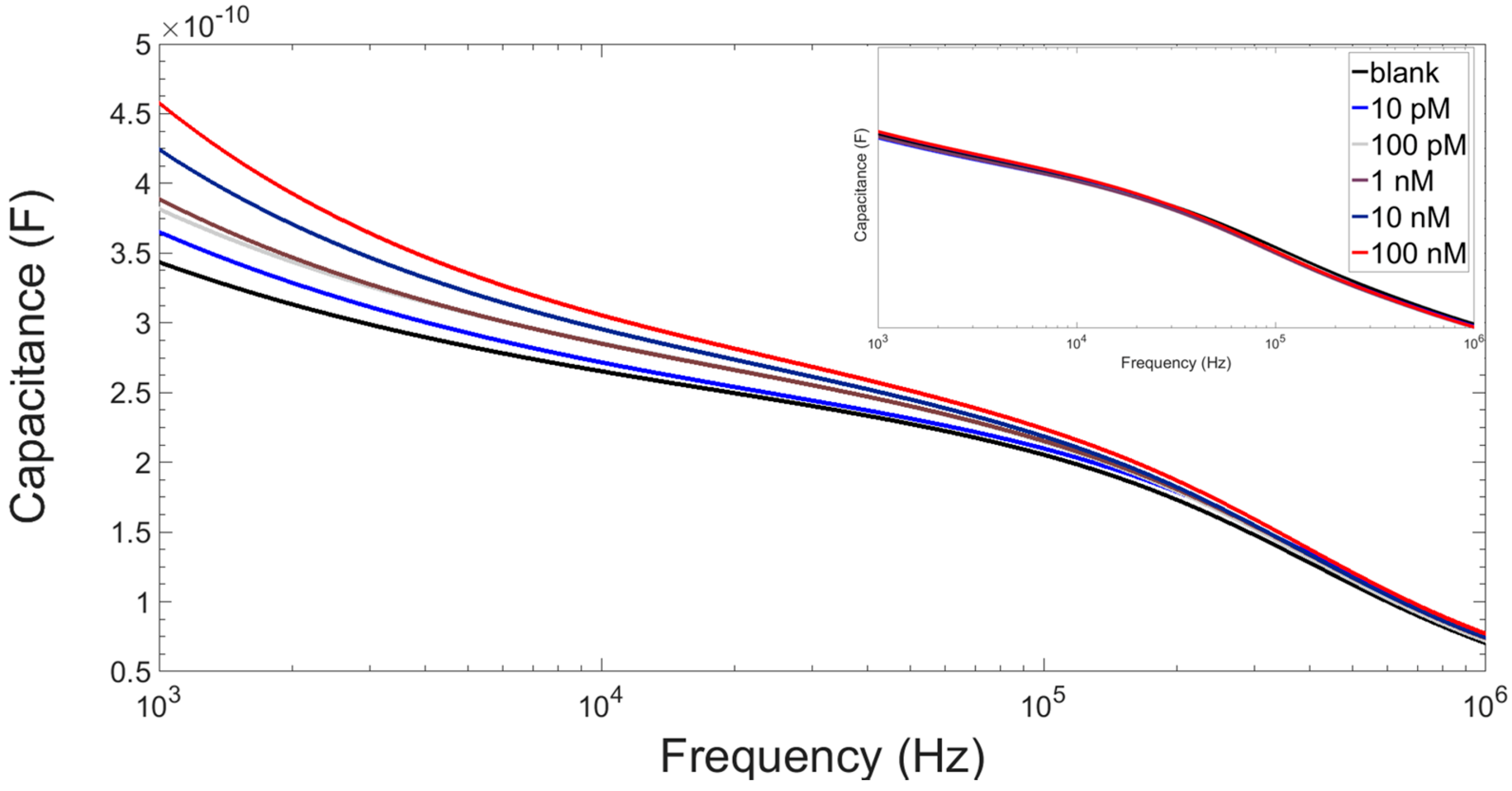
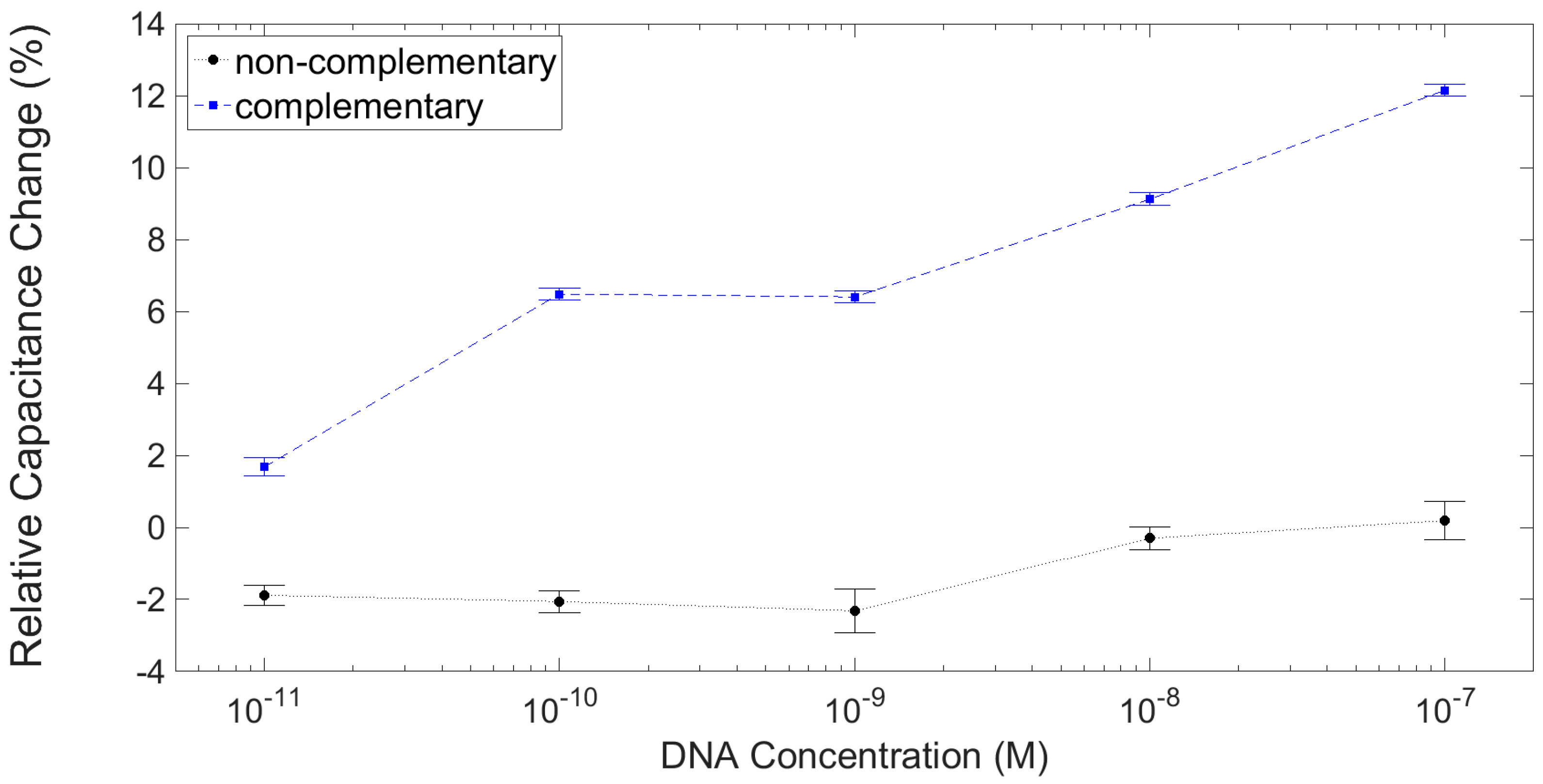
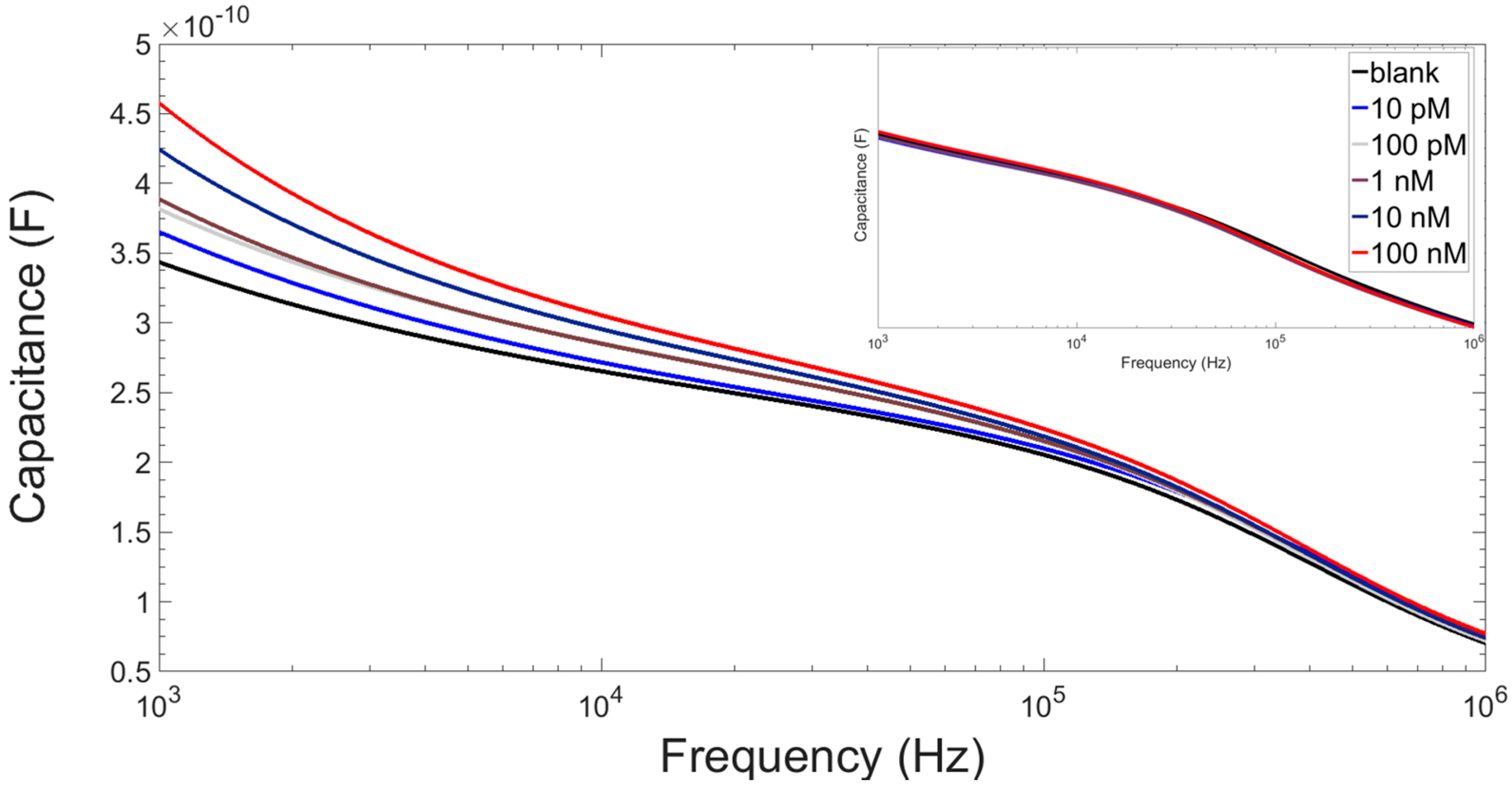
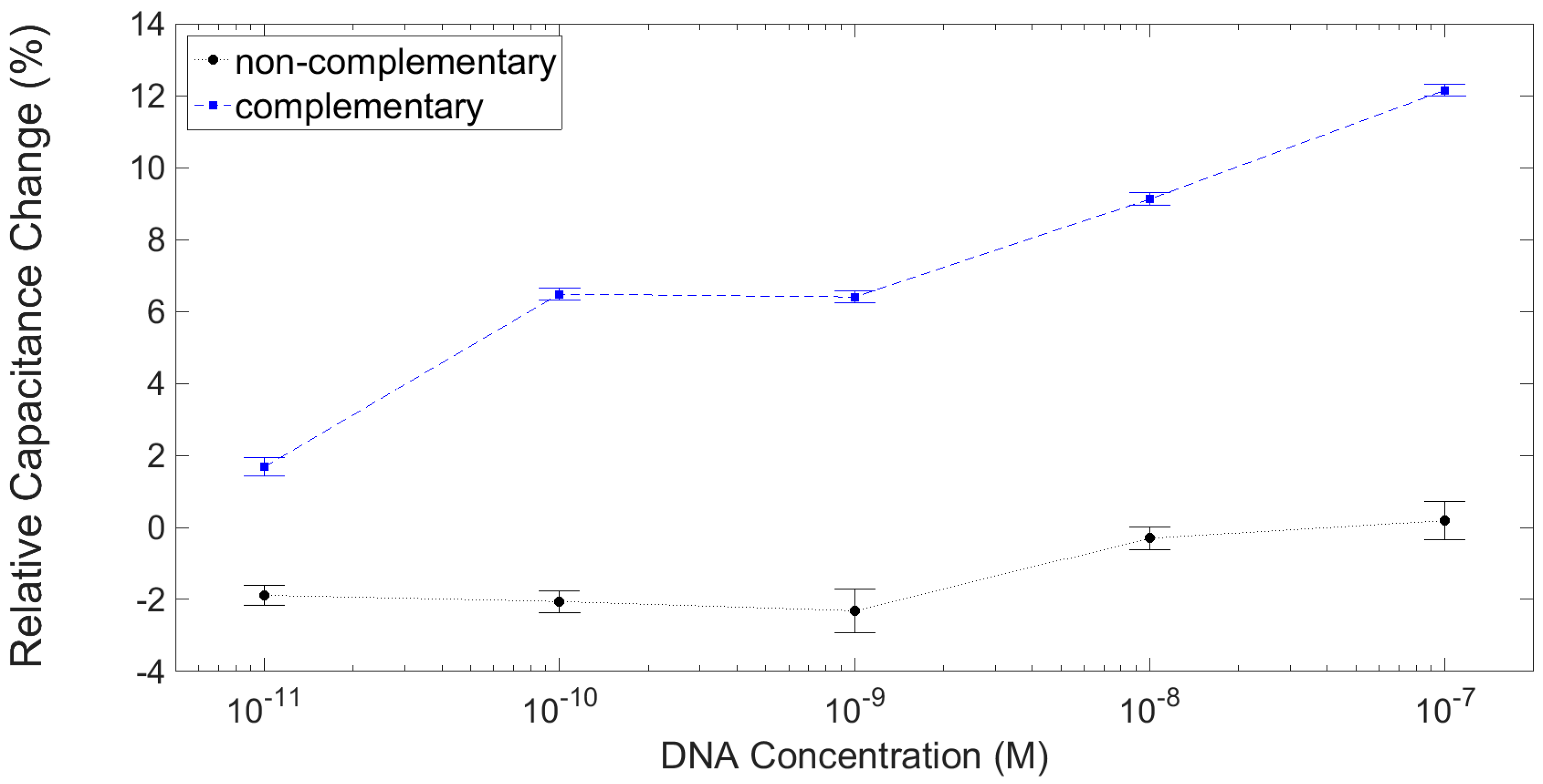
3.3. Results
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Weimann, T.; Hinze, P.; Scherer, H.; Niemeyer, J. Fabrication of a metallic single electron tunneling transistor by multilayer technique using lithography with a scanning transmission electron microscope. Microelectron. Eng. 1999, 46, 165–168. [Google Scholar] [CrossRef]
- Guillorn, M.A.; Carr, D.W.; Tiberio, R.C.; Greenbaum, E.; Simpson, M.L. Fabrication of dissimilar metal electrodes with nanometer interelectrode distance for molecular electronic device characterization. J. Vac. Sci. Technol. B 2000, 18, 1177–1181. [Google Scholar] [CrossRef]
- Reed, M.A.; Zhou, C.; Muller, C.J.; Burgin, T.P.; Tour, J.M. Conductance of a Molecular Junction. Science 1997, 278, 252–254. [Google Scholar] [CrossRef]
- Nagase, T.; Kubota, T.; Mashiko, S. Fabrication of nano-gap electrodes for measuring electrical properties of organic molecules using a focused ion beam. Thin Solid Films 2003, 438–439, 374–377. [Google Scholar] [CrossRef]
- Cui, A.; Liu, Z.; Dong, H.; Wang, Y.; Zhen, Y.; Li, W.; Li, J.; Gu, C.; Hu, W. Single Grain Boundary Break Junction for Suspended Nanogap Electrodes with Gapwidth Down to 1–2 nm by Focused Ion Beam Milling. Adv. Mater. 2015, 27, 3002–3006. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Myers, M.; Xiao, S.; Lefenfeld, M.; Steiner, R.; Tulevski, G.S.; Tang, J.; Baumert, J.; Leibfarth, F.; Yardley, J.T.; et al. Chemoresponsive monolayer transistors. Proc. Natl. Acad. Sci. USA 2006, 103, 11452–11456. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Pasupathy, A.N.; Goldsmith, J.I.; Chang, C.; Yaish, Y.; Petta, J.R.; Rinkoski, M.; Sethna, J.P.; Abruña, H.D.; McEuen, P.L.; et al. Coulomb blockade and the Kondo effect in single-atom transistors. Nature 2002, 417, 722–725. [Google Scholar] [CrossRef] [PubMed]
- Ito, M.; Morihara, K.; Toyonaka, T.; Takikawa, K.; Shirakashi, J.I. High-throughput nanogap formation by field-emission-induced electromigration. J. Vac. Sci. Technol. B 2015, 33, 051801. [Google Scholar] [CrossRef]
- Morpurgo, A.F.; Marcus, C.M.; Robinson, D.B. Controlled fabrication of metallic electrodes with atomic separation. Appl. Phys. Lett. 1999, 74, 2084–2086. [Google Scholar] [CrossRef]
- Hatzor, A.; Weiss, P. Molecular rulers for scaling down nanostructures. Science 2001, 291, 1019–1020. [Google Scholar] [PubMed]
- Lee, C.; Yang, E.H.; Myung, N.V.; George, T. A Nanochannel Fabrication Technique without Nanolithography. Nano Lett. 2003, 3, 1339–1340. [Google Scholar] [CrossRef]
- Kim, J.H.; Moon, H.; Yoo, S.; Choi, Y.K. Nanogap Electrode Fabrication for a Nanoscale Device by Volume-Expanding Electrochemical Synthesis. Small 2011, 7, 2210–2216. [Google Scholar] [CrossRef] [PubMed]
- Jain, T.; Tang, Q.; Bjørnholm, T.; Nørgaard, K. Wet Chemical Synthesis of Soluble Gold Nanogaps. Acc. Chem. Res. 2014, 47, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Piner, R.D.; Zhu, J.; Xu, F.; Hong, S.; Mirkin, C.A. “Dip-Pen” Nanolithography. Science 1999, 283, 661–663. [Google Scholar] [CrossRef] [PubMed]
- Fan, F.R.F.; Bard, A.J. Electrochemical detection of single molecules in zeptoliter volumes. Science 1995, 267, 871–874. [Google Scholar] [CrossRef] [PubMed]
- Pourhossein, P.; Chiechi, R.C. Fabricating Nanogaps by Nanoskiving. J. Vis. Experiments 2013. [Google Scholar] [CrossRef] [PubMed]
- Strobel, S.; Arinaga, K.; Hansen, A.; Tornow, M. A silicon-on-insulator vertical nanogap device for electrical transport measurements in aqueous electrolyte solution. Nanotechnology 2007, 18, 295201–295205. [Google Scholar] [CrossRef]
- Jang, D.Y.; Kim, Y.P.; Kim, H.S.; Park, S.H.K.; Choi, S.Y.; Choi, Y.K. Sublithographic vertical gold nanogap for label-free electrical detection of protein-ligand binding. J. Vac. Sci. Technol. B 2007, 25, 443–447. [Google Scholar] [CrossRef]
- Wolfrum, B.; Zevenbergen, M.; Lemay, S. Nanofluidic Redox Cycling Amplification for the selective detection of Catechol. Anal. Chem. 2008, 80, 972–977. [Google Scholar] [CrossRef] [PubMed]
- Zevenbergen, M.A.G.; Singh, P.S.; Goluch, E.D.; Wolfrum, B.L.; Lemay, S.G. Stochastic Sensing of Single Molecules in a Nanofluidic Electrochemical Device. Nano Lett. 2011, 11, 2881–2886. [Google Scholar] [CrossRef] [PubMed]
- Schukfeh, M.I.; Sepunaru, L.; behr, P.; Li, W.; Pecht, I.; Sheves, M.; Cahen, D.; Tornow, M. Towards nanometer-spaced silicon contacts to proteins. Nanotechnology 2016, 27, 115302. [Google Scholar] [CrossRef] [PubMed]
- Krahne, R.; Yacoby, A.; Shtrikman, H.; Bar-Joseph, I.; Dadosh, T.; Sperling, J. Fabrication of nanoscale gaps in integrated circuits. Appl. Phys. Lett. 2002, 81, 730–732. [Google Scholar] [CrossRef]
- McCreery, R.L.; Bergren, A.J. Progress with Molecular Electronic Junctions: Meeting Experimental Challenges in Design and Fabrication. Adv. Mater. 2009, 21, 4303–4322. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Hu, W.; Zhu, D. Nanogap Electrodes. Adv. Mater. 2010, 22, 286–300. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Guo, Z.; Yang, G.M.; Li, J.; Li, M.Q.; Liu, J.H.; Huang, X.J. Electrical nanogap devices for biosensing. Mater. Today 2010, 13, 28–41. [Google Scholar] [CrossRef]
- Rassaei, L.; Mathwig, K.; Kang, S.; Heering, H.A.; Lemay, S.G. Integrated Biodetection in a Nanofluidic Device. ACS Nano 2014, 8, 8278–8284. [Google Scholar] [CrossRef] [PubMed]
- Kätelhön, E.; Mayer, D.; Banzet, M.; Offenhäusser, A.; Wolfrum, B. Nanocavity crossbar arrays for parallel electrochemical sensing on a chip. Beilstein J. Nanotechnol. 2014, 5, 1137–1143. [Google Scholar] [CrossRef] [PubMed]
- Hüske, M.; Stockmann, R.; Offenhäusser, A.; Wolfrum, B. Redox cycling in a nanoporous electrochemical devices. Nanoscale 2014, 6, 589–598. [Google Scholar] [CrossRef] [PubMed]
- Wolfrum, B.; Kätelhön, E.; Yakushenko, A.; Krause, K.J.; Adly, N.; Hüske, M.; Rinklin, P. Nanoscale Electrochemical Sensor Arrays: Redox Cycling Amplification In Dual-Electrode Systems. Acc. Chem. Res. 2016, 49, 2031–2040. [Google Scholar] [CrossRef] [PubMed]
- Goluch, E.D.; Wolfrum, B.; Singh, P.S.; Zevenbergen, M.A.G.; Lemay, S.G. Redox cycling in nanofluidic channels using interdigitated electrodes. Anal. Bioanal. Chem. 2009, 394, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Hatsuki, R.; Yujiro, F.; Yamamoto, T. Direct measurement of electric double layer in a nanochannel by electrical impedance spectroscopy. Microfluid. Nanofluid. 2013, 14, 983–988. [Google Scholar] [CrossRef]
- Hatsuki, R.; Honda, A.; Kajitani, M.; Yamamoto, T. Nonlinear electrical impedance spectroscopy of viruses using very high electric fields created by nanogap electrodes. Front. Microbiol. 2015, 6, 940. [Google Scholar] [CrossRef] [PubMed]
- Hsueh, H.T.; Lin, C.T. An incremental double-layer capacitance of a planar nano gap and its application in cardiac-troponin T detection. Biosens. Bioelectron. 2016, 79, 636–643. [Google Scholar] [CrossRef] [PubMed]
- Ionescu-Zanetti, C.; Nevill, J.T.; Di Carlo, D.; Jeong, K.H.; Lee, L.P. Nanogap capacitors: Sensitivity to sample permittivity changes. J. Appl. Phys. 2006, 99, 024305. [Google Scholar] [CrossRef]
- Kanno, Y.; Ino, K.; Shiku, H.; Matsue, T. A local redox cycling-based electrochemical chip device with nanocavities for multi-electrochemical evaluation of embryoid bodies. Lab Chip 2015, 15, 4404–4414. [Google Scholar] [CrossRef] [PubMed]
- Penchev, P.; Shang, X.; Dimov, S.; Lancaster, M. Novel Manufacturing Route for Scale up Production of Terahertz Technology Devices. J. Micro Nano-Manuf. 2016, 4, 021002. [Google Scholar] [CrossRef]
- Lee, T.M.H.; Lee, D.H.Y.; Liaw, C.Y.N.; Lao, A.I.K.; Hsing, L.I.M. Detailed characterization of anodic bonding process between glass and thin-film coated silicon substrates. Sens. Actuators A Phys. 2000, 86, 103–107. [Google Scholar] [CrossRef]
- Chen, M.X.; Yi, X.J.; Gan, Z.Y.; Liu, S. Reliability of anodically bonded silicon—glass packages. Sens. Actuators A Phys. 2005, 120, 291–295. [Google Scholar] [CrossRef]
- He, J.; Yang, F.; Wang, W.; Zhang, L.; Huang, X.; Zhang, D. Electric current characteristics of anodic bonding. J. Micromech. Microeng. 2015, 25, 065002. [Google Scholar] [CrossRef]
- Shakeel, S.; Karim, S.; Ali, A. Peptide nucleic acid (PNA)—A review. J. Chem. Technol. Biotechnol. 2006, 81, 892–899. [Google Scholar] [CrossRef]
- Oleinikova, A.; Sasisanker, P.; Weingärtner, H. What Can Really Be Learned from Dielectric Spectroscopy of Protein Solutions? A Case Study of Ribonuclease A. J. Phys. Chem. B. 2004, 108, 8467–8474. [Google Scholar] [CrossRef]
- Yi, M.; Jeong, K.H.; Lee, L.P. Theoretical and experimental study towards a nanogap dielectric biosensor. Biosens. Bioelectron. 2005, 20, 1320–1326. [Google Scholar] [CrossRef] [PubMed]
- Mannoor, M.S.; James, T.; Ivanov, D.V.; Beadling, L.; Braunlin, W. Nanogap Dielectric Spectroscopy for Aptamer-Based Protein Detection. Biophys. J. 2010, 98, 724–732. [Google Scholar] [CrossRef] [PubMed]
- Shields, P.A.; Allsopp, D.W.E. Nanoimprint lithography resist profile inversion for lift-off applications. Microelectron. Eng. 2011, 88, 3011–3014. [Google Scholar] [CrossRef]
- Hammond, J.L.; Gross, A.J.; Estrela, P.; Iniesta, J.; Green, S.J.; Winlove, C.P.; Benjamin, N.; Marken, F. Cysteine-cystine redox cycling in a gold-gold dual-plate generator-collector microtrench sensor. Anal. Chem. 2014, 86, 6748–6752. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fabrication | Type | Orientation | Electrode | Gap | Area | Reference |
|---|---|---|---|---|---|---|
| EBL + SE (Cr) | Nanofluidic linear IDA | H | Pt | ~250 nm | ~1.4 × 10−9 m2 | Goluch et al. [30] |
| ES | Parallel plate | H | Pt | ~50 nm | ~2.0 × 10−12 m2 | Kim et al. [12] |
| FIB | Parallel plate | H | Au | ~60 nm | ~1.6 × 10−13 m2 | Hatsuki et al. [31] |
| FIB | Parallel plate | H | Au | ~510 nm | ~2.5 × 10−12 m2 | Hatsuki et al. [32] |
| EBL | Parallel plate | H | Au | ~200 nm | ~2.4 × 10−12 m2 | Hsueh et al. [33] |
| EBL | Serpentine parallel plate | H | Au | ~215 nm | ~3.3 × 10−9 m2 | This work |
| SE (SiO2) | Nanocavity linear array | V | Si | ~90 nm | ~2.1 × 10−7 m2 | Ionescu-Zanetti et al. [34] |
| SE (SiO2) + AD | Nanocavity | V | Au | ~65 nm | ~2.0 × 10−13 m2 | Strobel et al. [17] |
| SE (Cr) | Nanofluidic parallel plate cavity | V | Pt | ~70 nm | ~1.5 × 10−10 m2 | Zevenbergen et al. [20] |
| SE (Cr) | Nanocavity crossbar array | V | Pt | ~65 nm | ~1.7 × 10−12 m2 | Kätelhön et al. [27] |
| SE (Cr) | Nanofluidic parallel plate cavity | V | Au | ~200 nm | ~3.0 × 10−11 m2 | Rassaei et al. [26] |
| EBL + SE (Si3N4) | Nanoporous ring-ring array | V | Pt | ~100 nm | ~3.6 × 10−9 m2 | Hüske et al. [28] |
| SE (Cr) | Nanocavity ring-ring array | V | Pt | ~230 nm | 2.7 × 10−7 m2 | Kanno et al. [35] |
| ICP RIE + AB | Parallel plate | V | Au | ~500 nm | 3.0 × 10−6 m2 | This work |
| Parameter | Value |
|---|---|
| SF6 | 50 SCCM |
| O2 | 8 SCCM |
| RF | 90 W |
| ICP | 8 s 1300 W 1, 32 s 1200 W |
| Pressure | 10 mTorr |
| He backing | 5 SCCM |
| Temperature | 0.0 °C |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hammond, J.L.; Rosamond, M.C.; Sivaraya, S.; Marken, F.; Estrela, P. Fabrication of a Horizontal and a Vertical Large Surface Area Nanogap Electrochemical Sensor. Sensors 2016, 16, 2128. https://doi.org/10.3390/s16122128
Hammond JL, Rosamond MC, Sivaraya S, Marken F, Estrela P. Fabrication of a Horizontal and a Vertical Large Surface Area Nanogap Electrochemical Sensor. Sensors. 2016; 16(12):2128. https://doi.org/10.3390/s16122128
Chicago/Turabian StyleHammond, Jules L., Mark C. Rosamond, Siva Sivaraya, Frank Marken, and Pedro Estrela. 2016. "Fabrication of a Horizontal and a Vertical Large Surface Area Nanogap Electrochemical Sensor" Sensors 16, no. 12: 2128. https://doi.org/10.3390/s16122128
APA StyleHammond, J. L., Rosamond, M. C., Sivaraya, S., Marken, F., & Estrela, P. (2016). Fabrication of a Horizontal and a Vertical Large Surface Area Nanogap Electrochemical Sensor. Sensors, 16(12), 2128. https://doi.org/10.3390/s16122128