
Optimized Detection of Plasmodium falciparum Topoisomerase I Enzyme Activity in a Complex Biological Sample by the Use of Molecular Beacons

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
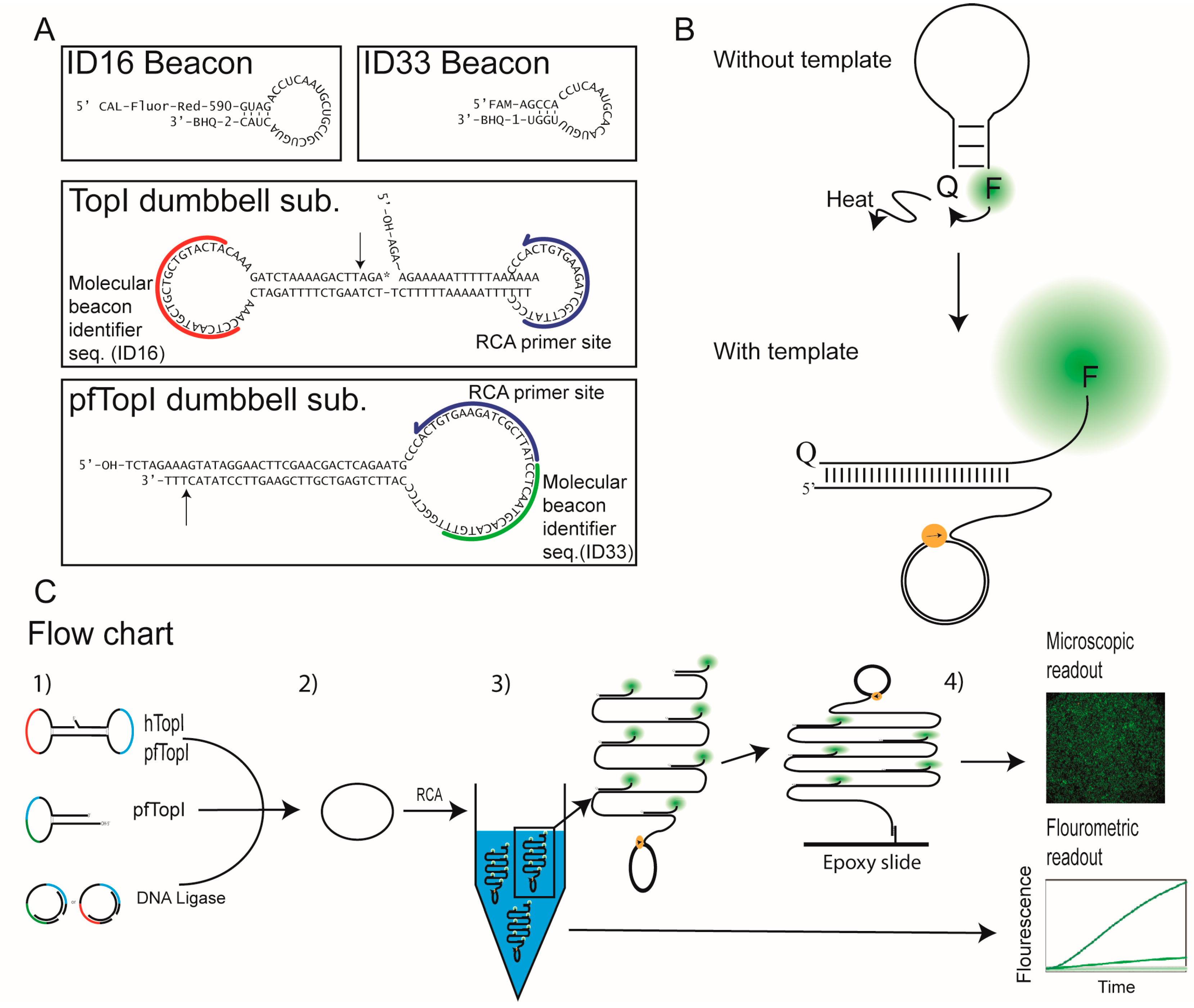
2.1. Oligonucleotides, Substrates and Molecular Beacons
2.1.1. Oligonucleotides for Preparation of c.c.
2.1.2. Dumbbell Substrates
2.1.3. Molecular Beacons
2.2. Preparation of Control Circles
2.3. REEAD Experiments
2.4. RCA and Fluorometric Analysis
2.5. Microscopic Analysis
2.6. Expression and Purification Enzymes
2.7. Preparation of Cell Extracts
2.8. Data Processing
3. Results
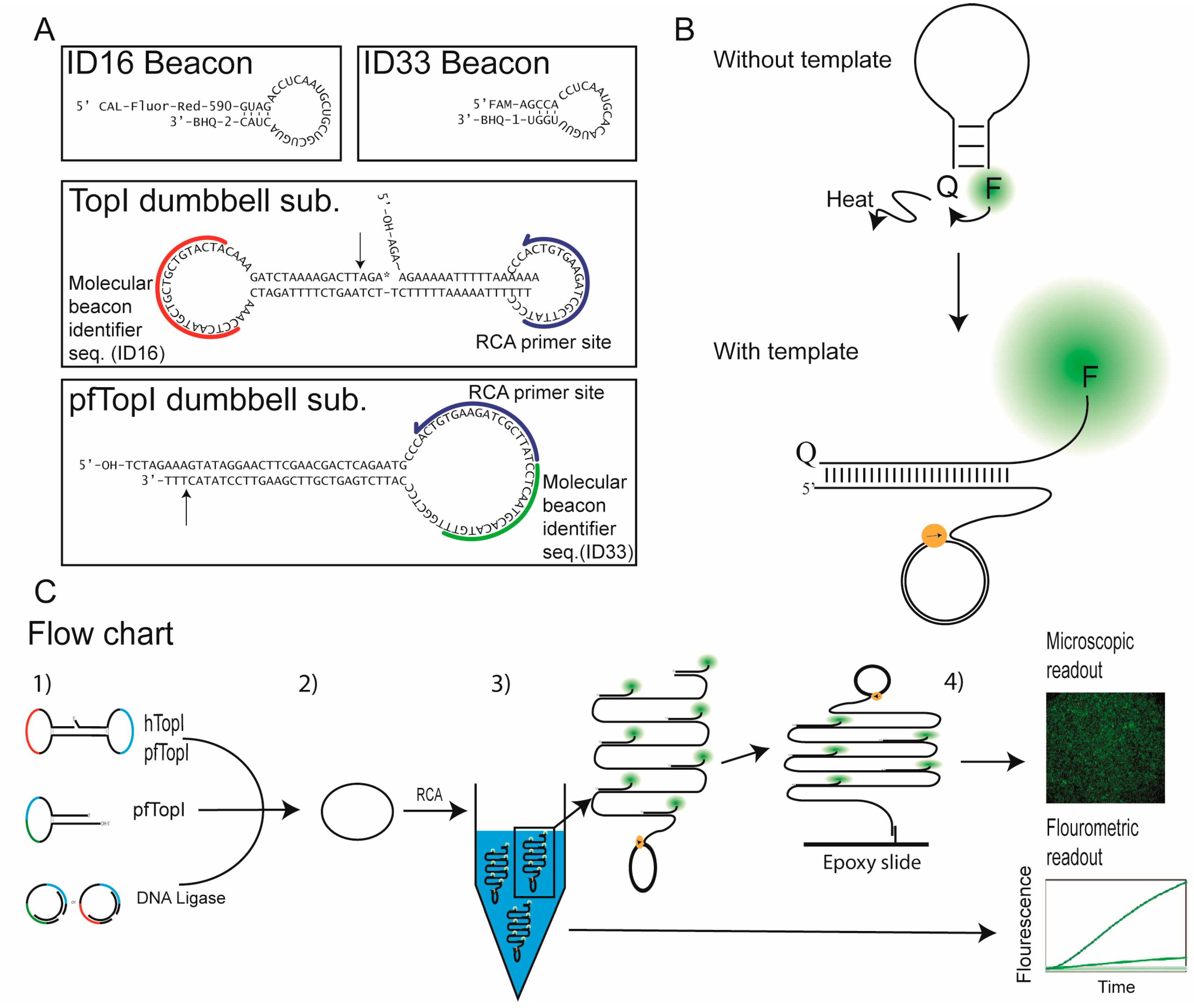
3.1. Design of the Molecular Beacons and REEAD Setup
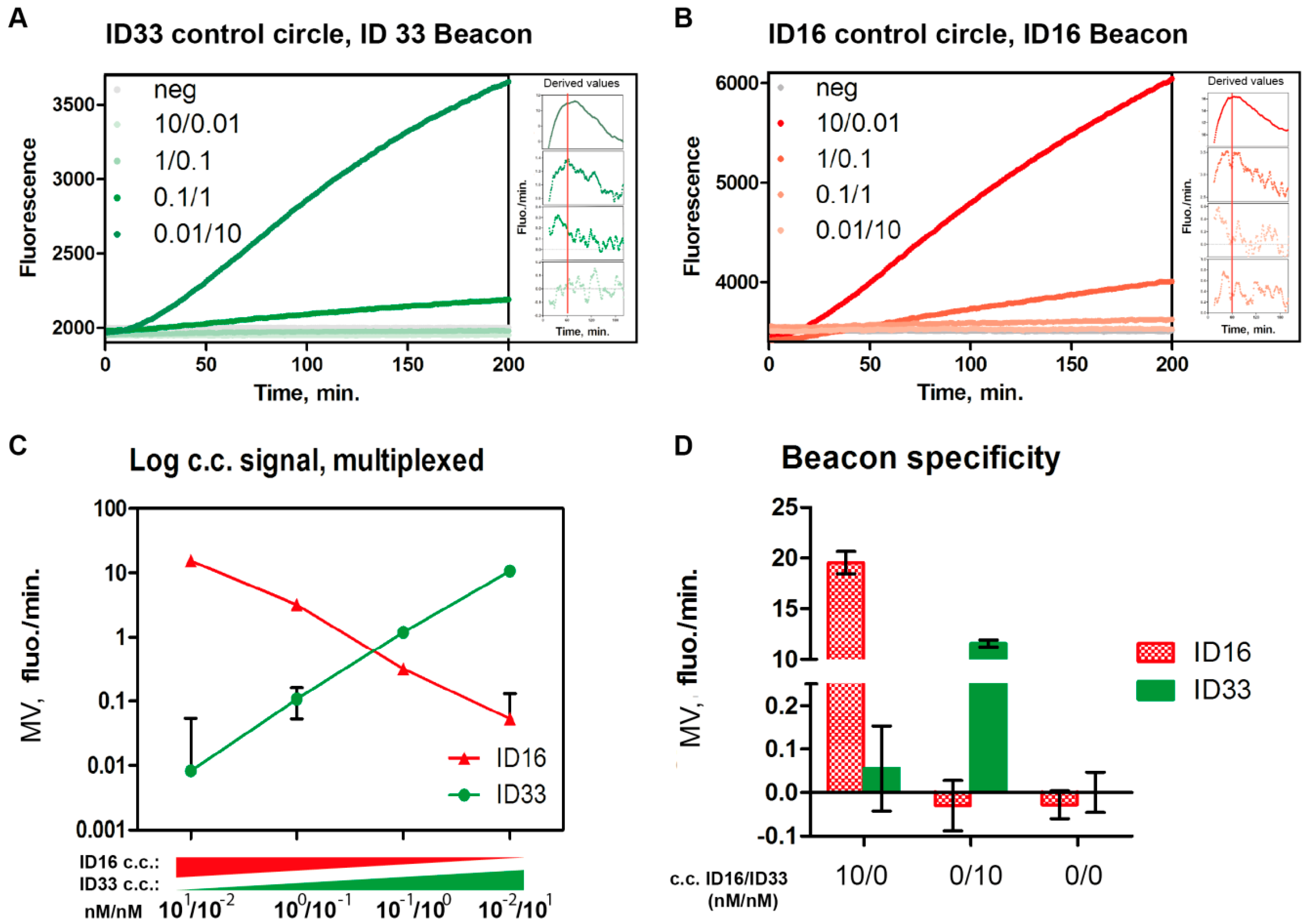
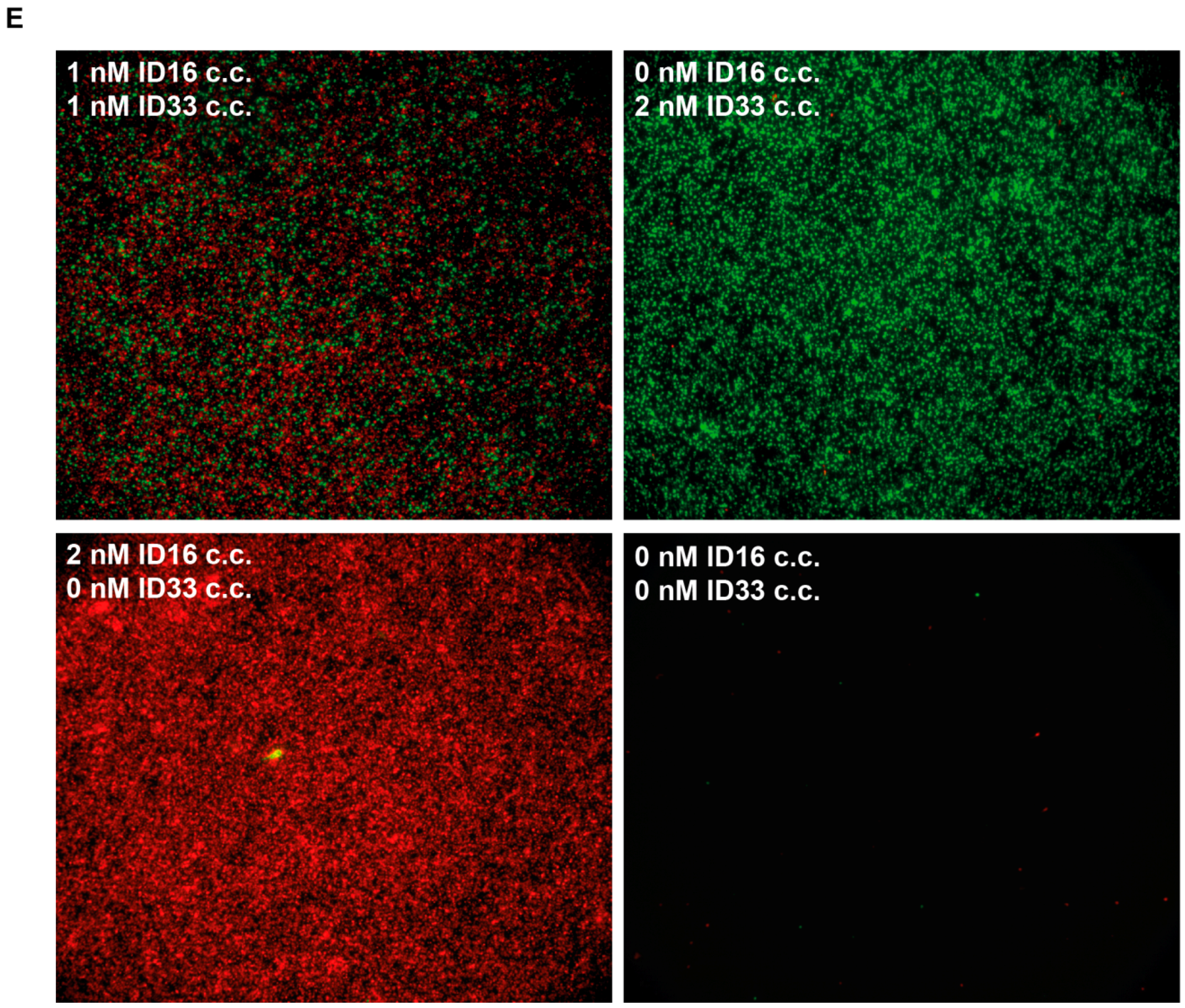
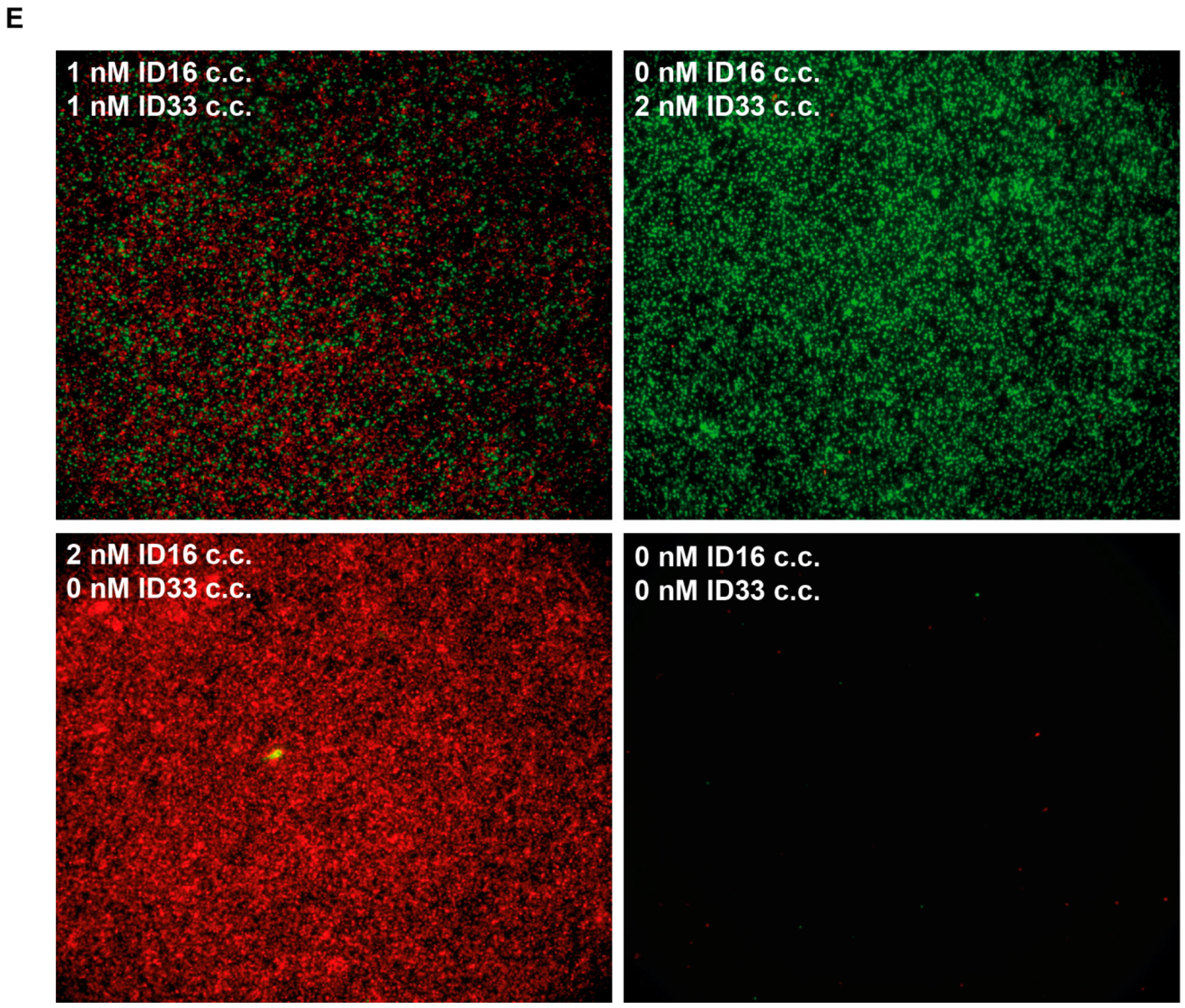
3.2. The Use of Molecular Beacons for Readout Allows for Multiplexed Detection of Two RCA Reactions Performed in the Same Test Tube
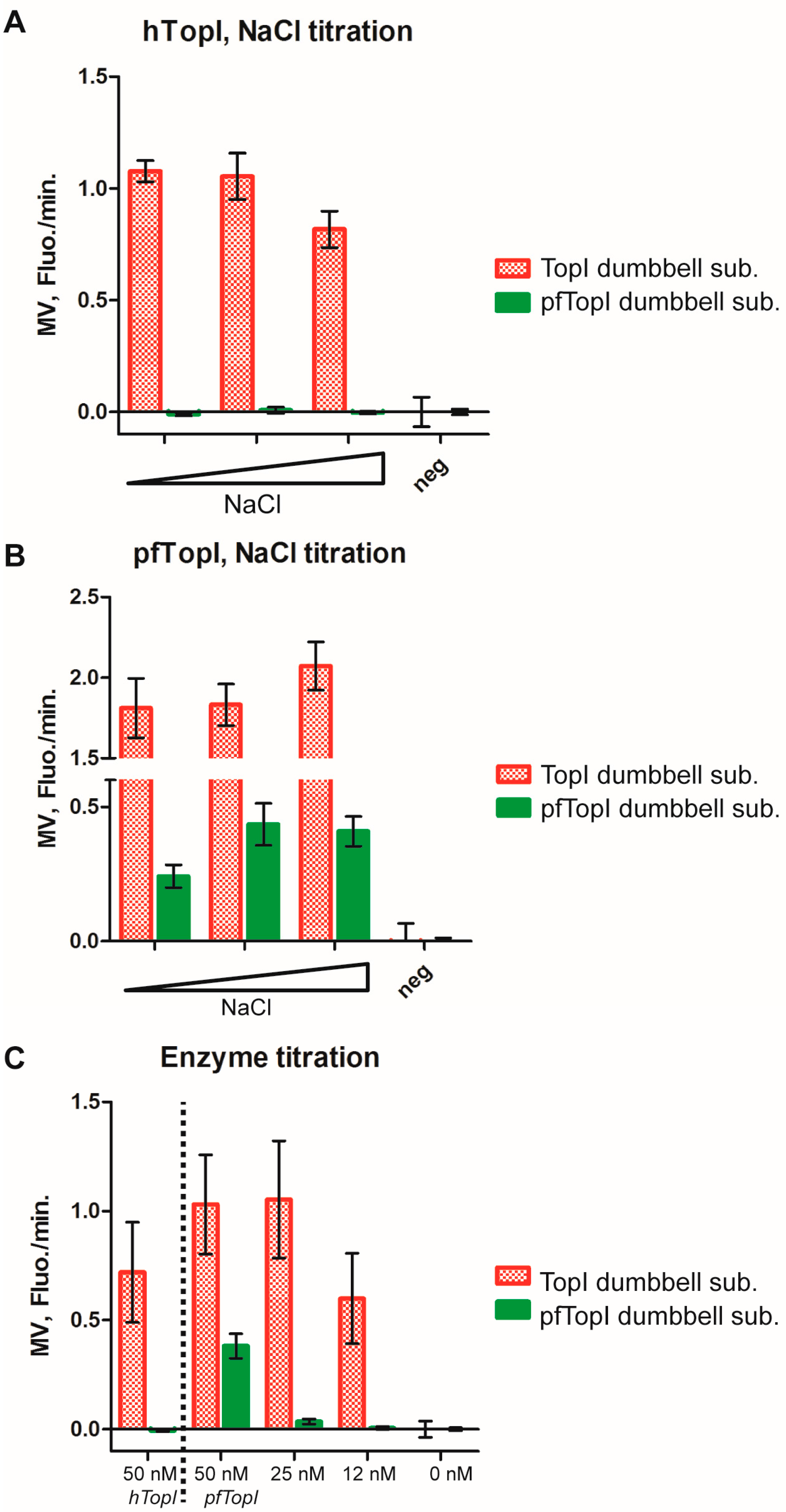
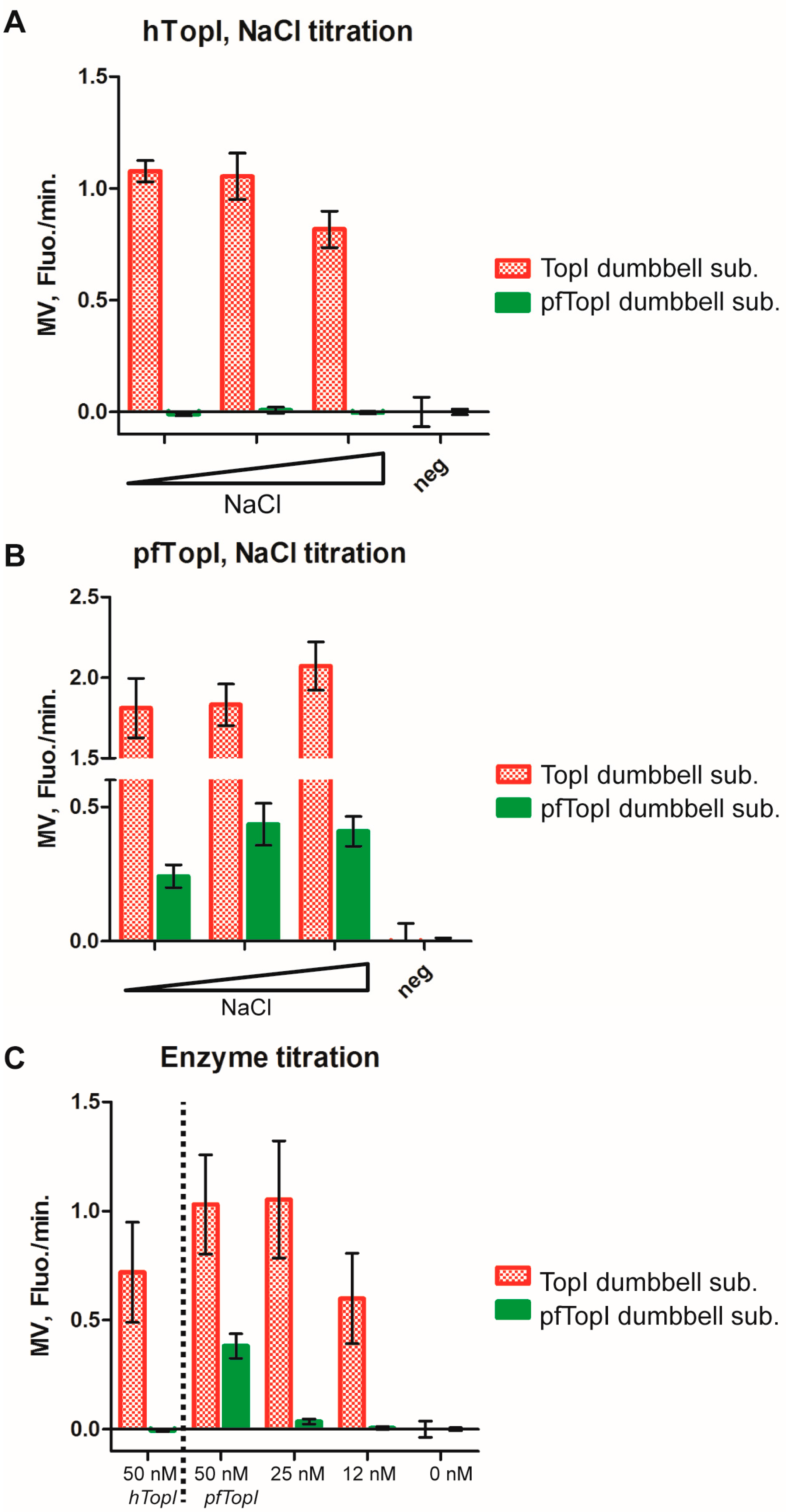
3.3. Molecular Beacon Based Real-Time Detection of RCA Performed on TopI Generated DNA Circles
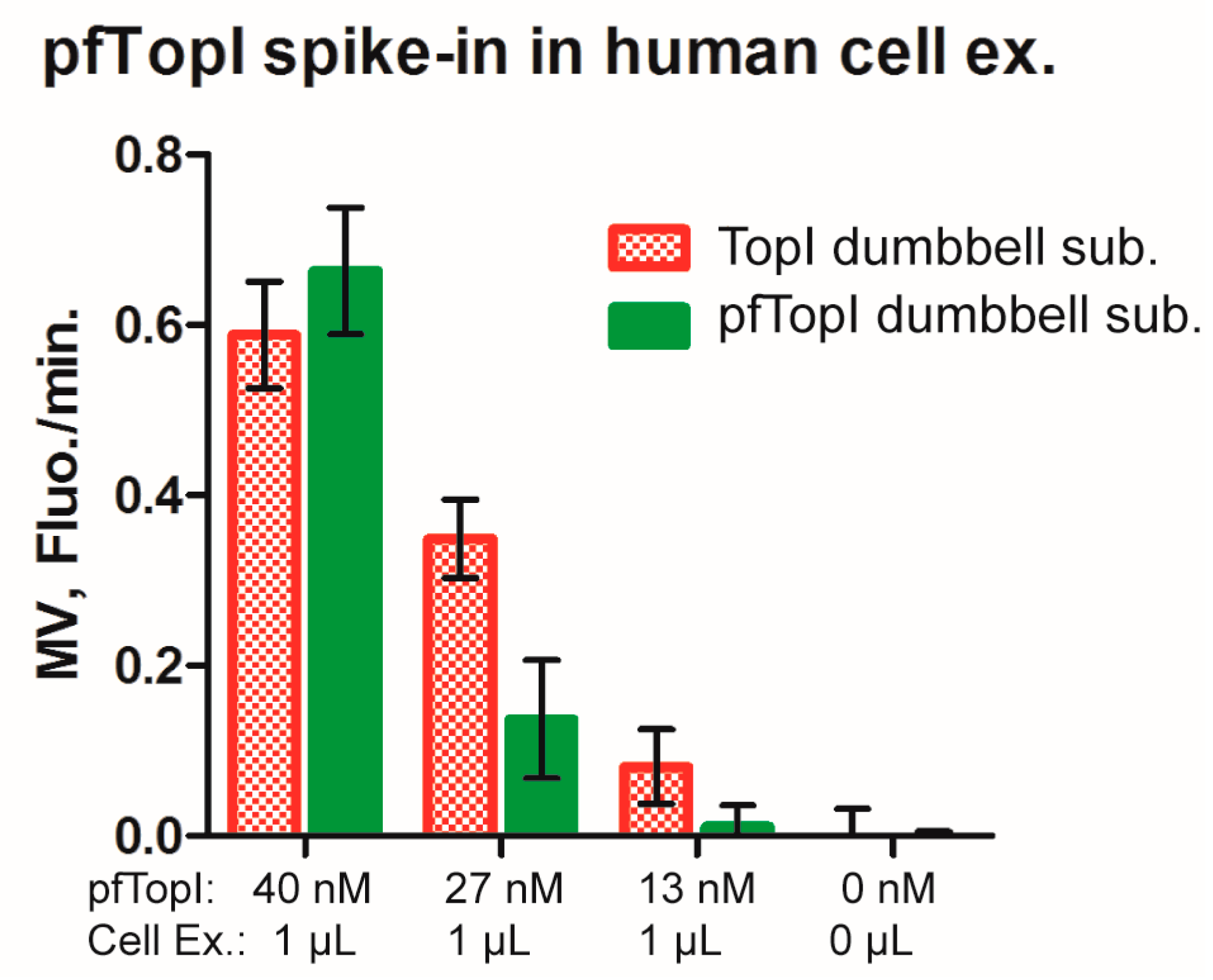
3.4. Multiplexed Detection of Enzyme Activities from hTopI and pfTopI in the Presence of Complex Human Samples
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kettling, U.; Koltermann, A.; Schwille, P.; Eigen, M. Real-time enzyme kinetics monitored by dual-color fluorescence cross-correlation spectroscopy. Proc. Natl. Acad. Sci. USA 1998, 95, 1416–1420. [Google Scholar] [CrossRef] [PubMed]
- Hardin, A.H.; Sarkar, S.K.; Seol, Y.; Liou, G.F.; Osheroff, N.; Neuman, K.C. Direct measurement of DNA bending by type IIA topoisomerases: Implications for non-equilibrium topology simplification. Nucleic Acids Res. 2011, 39, 5729–5743. [Google Scholar] [CrossRef] [PubMed]
- Kristoffersen, E.L.; Jørgensen, L.A.; Franch, O.; Etzerodt, M.; Frøhlich, R.; Bjergbæk, L.; Stougaard, M.; Ho, Y.P.; Knudsen, B.R. Real-time investigation of human topoisomerase I reaction kinetics using an optical sensor: A fast method for drug screening and determination of active enzyme concentrations. Nanoscale 2015, 7, 9825–9834. [Google Scholar] [CrossRef] [PubMed]
- Jensen, P.W.; Falconi, M.; Kristoffersen, E.L.; Simonsen, A.T.; Cifuentes, J.B.; Marcussen, L.B.; Frøhlich, R.; Vagner, J.; Harmsen, C.; Juul, S.; et al. Real-time detection of TDP1 activity using a fluorophore-quencher coupled DNA-biosensor. Biosens. Bioelectron. 2013, 48C, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Stougaard, M.; Lohmann, J.S.; Mancino, A.; Celik, S.S.; Andersen, F.F.; Koch, J.J. Single-Molecule Detection of Human Topoisomerase I Cleavage-Ligation Activity. ACS Nano 2009, 3, 223–233. [Google Scholar] [CrossRef] [PubMed]
- Marcussen, L.B.; Lohmann, J.S.; Mancino, A.; Celik, S.; Andersen, F.F.; Koch, J.; Knudsen, B.R. DNA-based sensor for real-time measurement of the enzymatic activity of human topoisomerase I. Sensors 2013, 13, 4017–4028. [Google Scholar] [CrossRef] [PubMed]
- Andersen, F.F.; Stougaard, M.; Jørgensen, H.L.; Bendsen, S.; Juul, S.; Hald, K.; Andersen, A.H.; Koch, J.; Knudsen, B.R. Multiplexed detection of site specific recombinase and DNA topoisomerase activities at the single molecule level. ACS Nano 2009, 3, 4043–4054. [Google Scholar] [CrossRef] [PubMed]
- Tesauro, C.; Juul, S.; Arnò, B.; Nielsen, C.J.; Fiorani, P.; Frøhlich, R.F.; Andersen, F.F.; Desideri, A.; Stougaard, M.; Petersen, E.; et al. Specific detection of topoisomerase I from the malaria causing P. falciparum parasite using isothermal Rolling Circle Amplification. In Proceedings of the 2012 Annual International Conference of the IEEE Engineering in Medicine and Biology Society (EMBC), San Diego, CA, USA, 28 August–1 September 2012.
- Juul, S.; Nielsen, C.J.; Labouriau, R.; Roy, A.; Tesauro, C.; Jensen, P.W.; Harmsen, C.; Kristoffersen, E.L.; Chiu, Y.L.; Frøhlich, R.; et al. Droplet microfluidics platform for highly sensitive and quantitative detection of malaria-causing plasmodium parasites based on enzyme activity measurement. ACS Nano 2012, 6, 10676–10683. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Kang, T.S.; Lu, L.; Wang, W.; Ma, D.L.; Leung, C.H. A G-quadruplex-selective luminescent probe with an anchor tail for the switch-on detection of thymine DNA glycosylase activity. Biosens. Bioelectron. 2016, 86, 849–857. [Google Scholar] [CrossRef] [PubMed]
- Mullis, K.; Faloona, F.; Scharf, S.; Saiki, R.; Horn, G.; Erlich, H. Specific enzymatic amplification of DNA in vitro: The polymerase chain reaction. Cold Spring Harb. Symp. Quant. Biol. 1986, 51, 263–73. [Google Scholar] [CrossRef] [PubMed]
- White, T.B.; McCoy, A.M.; Streva, V.A.; Fenrich, J.; Deininger, P.L. A droplet digital PCR detection method for rare L1 insertions in tumors. Mob. DNA 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Xiong, E.; Zhang, X.; Liu, Y.; Zhou, J.; Yu, P.; Li, X.; Chen, J. Ultrasensitive Electrochemical Detection of Nucleic Acids Based on the Dual-Signaling Electrochemical Ratiometric Method and Exonuclease III-Assisted Target Recycling Amplification Strategy. Anal Chem. 2015, 87, 7291–7296. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Tan, T.; Chen, G.; Zhang, K.; Zhu, J.-J. Ultrasensitive electrochemical detection of BCR/ABL fusion gene fragment based on polymerase assisted multiplication coupling with quantum dot tagging. Electrochem. Commun. 2013, 35, 104–107. [Google Scholar] [CrossRef]
- Li, C.; Li, Y.; Xu, X.; Wang, X.; Chen, Y.; Yang, X.; Liu, F.; Li, N. Fast and quantitative differentiation of single-base mismatched DNA by initial reaction rate of catalytic hairpin assembly. Biosens. Bioelectron. 2014, 60, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.M.; Li, F.; Zhang, Z.; Kang, D.K.; Ankrum, J.A.; Le, X.C.; Zhao, W. Rolling circle amplification: A versatile tool for chemical biology, materials science and medicine. Chem. Soc. Rev. 2014, 43, 3324–3341. [Google Scholar] [CrossRef] [PubMed]
- Bi, S.; Zhao, T.; Luo, B.; Zhu, J.-J. Hybridization chain reaction-based branched rolling circle amplification for chemiluminescence detection of DNA methylation. Chem. Commun. 2013, 49, 6906–6908. [Google Scholar] [CrossRef] [PubMed]
- Blanco, L.; Bernad, A.; Lazaro, J.M.; Martin, G.; Garmendia, C.; Salas, M. Highly efficient DNA synthesis by the phage Φ29 DNA polymerase. Symmetrical mode of DNA replication. J. Biol. Chem. 1989, 264, 8935–8940. [Google Scholar] [PubMed]
- Jiang, H.-X.; Liang, Z.-Z.; Ma, Y.-H.; Kong, D.-M.; Hong, Z.-Y. G-quadruplex fluorescent probe-mediated real-time rolling circle amplification strategy for highly sensitive microRNA detection. Anal. Chim. Acta 2016, 943, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.-X.; Zhao, M.-Y.; Niu, C.-D.; Kong, D.-M. Real-time monitoring of rolling circle amplification using aggregation-induced emission: Applications in biological detection. Chem. Commun. 2015, 51, 16518–16521. [Google Scholar] [CrossRef] [PubMed]
- Proszek, J.; Roy, A.; Jakobsen, A.K.; Frøhlich, R.; Knudsen, B.R.; Stougaard, M. Topoisomerase I as a biomarker: Detection of activity at the single molecule level. Sensors 2013, 14, 1195–1207. [Google Scholar] [CrossRef] [PubMed]
- Stougaard, M.; Juul, S.; Andersen, F.F.; Knudsen, B.R. Strategies for highly sensitive biomarker detection by Rolling Circle Amplification of signals from nucleic acid composed sensors. Integr. Biol. 2011, 3, 982–992. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Daubendiek, S.L.; Zillman, M.A.; Ryan, K.; Kool, E.T. Rolling Circle DNA Synthesis: Small Circular Oligonucleotides as Efficient Templates for DNA Polymerases. J. Am. Chem. Soc. 1996, 118, 1587–1594. [Google Scholar] [CrossRef] [PubMed]
- Fire, A.; Xu, S.Q. Rolling replication of short DNA circles. Proc. Natl. Acad. Sci. USA 1995, 92, 4641–4645. [Google Scholar] [CrossRef] [PubMed]
- Lizardi, P.M.; Huang, X.; Zhu, Z.; Bray-Ward, P.; Thomas, D.C.; Ward, D.C. Mutation detection and single-molecule counting using isothermal rolling-circle amplification. Nat. Genet. 1998, 19, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, S.; Strömberg, M.; Gómez de la Torre, T.Z.; Russell, C.; Gunnarsson, K.; Nilsson, M.; Svedlindh, P.; Strømme, M.; Leifer, K. Real-space transmission electron microscopy investigations of attachment of functionalized magnetic nanoparticles to DNA-coils acting as a biosensor. J. Phys. Chem. B 2010, 114, 13255–13262. [Google Scholar] [CrossRef] [PubMed]
- Blab, G.A.; Schmidt, T.; Nilsson, M. Homogeneous Detection of Single Rolling Circle Replication Products. Anal. Chem. 2004, 76, 495–498. [Google Scholar] [CrossRef] [PubMed]
- Cheglakov, Z.; Weizmann, Y.; Basnar, B.; Willner, I. Diagnosing viruses by the rolling circle amplified synthesis of DNAzymes. Org. Biomol. Chem. 2007, 5, 223–225. [Google Scholar] [CrossRef] [PubMed]
- Cheng, W.; Yan, F.; Ding, L.; Ju, H.; Yin, Y. Cascade signal amplification strategy for subattomolar protein detection by rolling circle amplification and quantum dots tagging. Anal. Chem. 2010, 82, 3337–3342. [Google Scholar] [CrossRef] [PubMed]
- Larsson, C.; Koch, J.; Nygren, A.; Janssen, G.; Raap, A.K.; Landegren, U.; Nilsson, M. In situ genotyping individual DNA molecules by target-primed rolling-circle amplification of padlock probes. Nat. Methods 2004, 1, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Mizuta, R.; Mizuta, M.; Kitamura, D. Atomic force microscopy analysis of rolling circle amplification of plasmid DNA. Arch. Histol. Cytol. 2003, 66, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Fung, C.W.; Cho, E.J.; Ellington, A.D. Real-Time Rolling Circle Amplification for Protein Detection. Anal. Chem. 2007, 79, 3320–3329. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, S.; Kramer, F. Molecular beacons: Probes that fluoresce upon hybridization. Nat. Biotechnol. 1996, 14, 303–308. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, M.; Gullberg, M.; Dahl, F.; Szuhai, K.; Raap, A.K. Real-time monitoring of rolling-circle amplification using a modified molecular beacon design. Nucleic Acids Res. 2002, 30, 1–7. [Google Scholar] [CrossRef]
- Kristoffersen, E.L.; Gonzalez, M.; Stougaard, M.; Tesauro, C. Molecular Beacon Enables Combination of Highly Processive and Highly Sensitive Rolling Circle Amplification Readouts for Detection of DNA-Modifying Enzymes. Nano Life 2015, 5. [Google Scholar] [CrossRef]
- Champoux, J.J. DNA topoisomerases: Structure, function, and mechanism. Annu. Rev. Biochem. 2001, 70, 369–413. [Google Scholar] [PubMed]
- Madden, K.R.; Champoux, J.J. Overexpression of Human Topoisomerase-I in Baby Hamster-Kidney Cells—Hypersensitivity of Clonal Isolates to Camptothecin. Cancer Res. 1992, 52, 525–532. [Google Scholar] [PubMed]
- Ikeguchi, M.; Arai, Y.; Maeta, Y.; Ashida, K.; Katano, K.; Wakatsuki, T. Topoisomerase I expression in tumors as a biological marker for CPT-11 chemosensitivity in patients with colorectal cancer. Surg. Today 2011, 41, 1196–1199. [Google Scholar] [CrossRef] [PubMed]
- Pommier, Y. Topoisomerase I inhibitors: Camptothecins and beyond. Nat. Rev. Cancer 2006, 6, 789–802. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Tesauro, C.; Frøhlich, R.; Hede, M.S.; Nielsen, M.J.; Kjeldsen, E.; Bonven, B.; Stougaard, M.; Gromova, I.; Knudsen, B.R. Decreased camptothecin sensitivity of the stem-cell-like fraction of Caco2 cells correlates with an altered phosphorylation pattern of topoisomerase I. PLoS ONE 2014, 9, e99628. [Google Scholar] [CrossRef] [PubMed]
- Stougaard, M.; Ho, Y.-P. DNA-based nanosensors for next-generation clinical diagnostics via detection of enzyme activity. Expert Rev. Mol. Diagn. 2014, 14, 1–3. [Google Scholar] [PubMed]
- Lisby, M.; Olesen, J.R.; Skouboe, C.; Krogh, B.O.; Straub, T.; Boege, F.; Velmurugan, S.; Martensen, P.M.; Andersen, A.H.; Jayaram, M.; et al. Residues within the N-terminal domain of human topoisomerase I play a direct role in relaxation. J. Biol. Chem. 2001, 276, 20220–20227. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; D’Annessa, I.; Nielsen, C.J.F.; Tordrup, D.; Laursen, R.R.; Knudsen, B.R.; Desideri, A.; Andersen, F.F. Peptide Inhibition of Topoisomerase IB from Plasmodium falciparum. Mol. Biol. Int. 2011, 2011, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Champoux, J.J. Proteins that affect DNA conformation. Annu. Rev. Biochem. 1978, 47, 449–479. [Google Scholar] [CrossRef] [PubMed]
- Carballeira, N.M.; Montano, N.; Alvarez-Velilla, R.; Prada, C.F.; Reguera, R.M.; Balaña-Fouce, R. Synthesis of marine α-methoxylated fatty acid analogs that effectively inhibit the topoisomerase IB from leishmania donovani with a mechanism different from that of camptothecin. Mar. Drugs 2013, 11, 3661–3675. [Google Scholar] [CrossRef] [PubMed]

© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Givskov, A.; Kristoffersen, E.L.; Vandsø, K.; Ho, Y.-P.; Stougaard, M.; Knudsen, B.R. Optimized Detection of Plasmodium falciparum Topoisomerase I Enzyme Activity in a Complex Biological Sample by the Use of Molecular Beacons. Sensors 2016, 16, 1916. https://doi.org/10.3390/s16111916
Givskov A, Kristoffersen EL, Vandsø K, Ho Y-P, Stougaard M, Knudsen BR. Optimized Detection of Plasmodium falciparum Topoisomerase I Enzyme Activity in a Complex Biological Sample by the Use of Molecular Beacons. Sensors. 2016; 16(11):1916. https://doi.org/10.3390/s16111916
Chicago/Turabian StyleGivskov, Asger, Emil L. Kristoffersen, Kamilla Vandsø, Yi-Ping Ho, Magnus Stougaard, and Birgitta R. Knudsen. 2016. "Optimized Detection of Plasmodium falciparum Topoisomerase I Enzyme Activity in a Complex Biological Sample by the Use of Molecular Beacons" Sensors 16, no. 11: 1916. https://doi.org/10.3390/s16111916
APA StyleGivskov, A., Kristoffersen, E. L., Vandsø, K., Ho, Y.-P., Stougaard, M., & Knudsen, B. R. (2016). Optimized Detection of Plasmodium falciparum Topoisomerase I Enzyme Activity in a Complex Biological Sample by the Use of Molecular Beacons. Sensors, 16(11), 1916. https://doi.org/10.3390/s16111916