
Colorimetric Integrated PCR Protocol for Rapid Detection of Vibrio parahaemolyticus

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Experimental
2.1. Chemicals and Reagents
2.2. Preparation of Magnetic Nanoparticles (MNPs) and Application in Genomic DNA Extraction of V. parahaemolyticus
2.3. PCR Amplification of V. parahaemolyticus
2.4. Colorimetric Detection of V. parahaemolyticus (33847 and 17802)
3. Results and Discussion
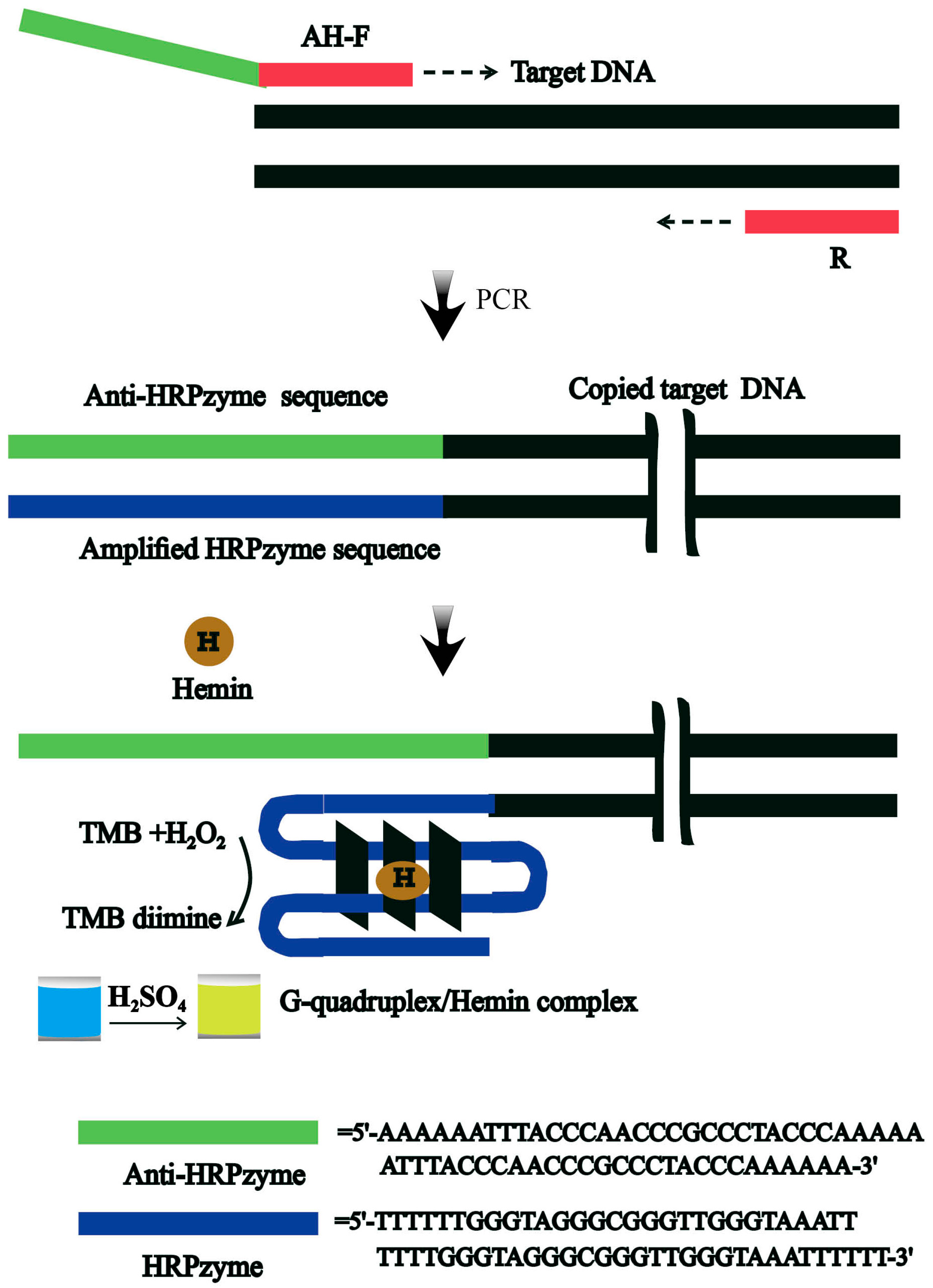
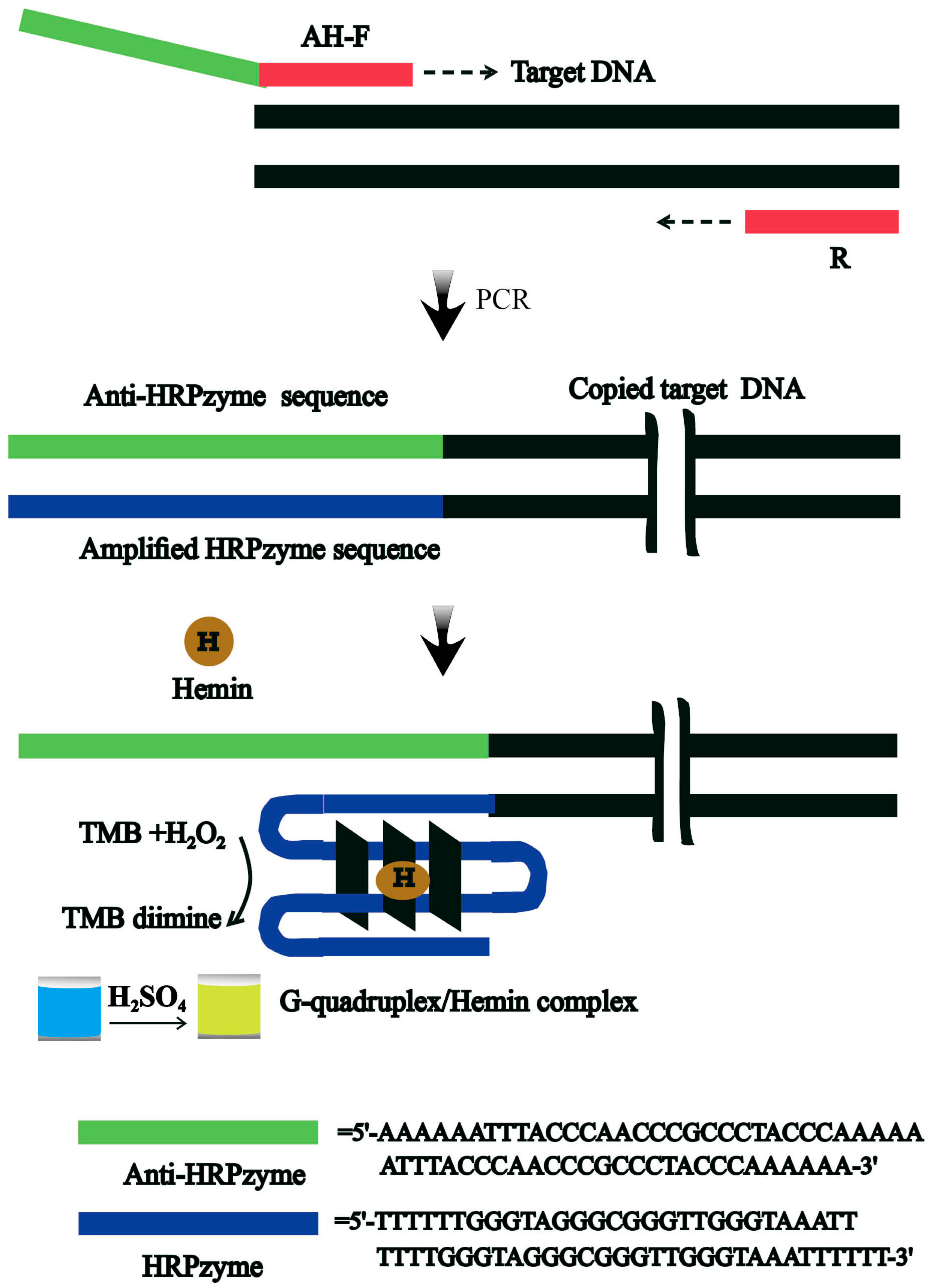
3.1. Design of the Rapid Colorimetric Strategy for Rapid and Easy Detection of V. parahaemolyticus
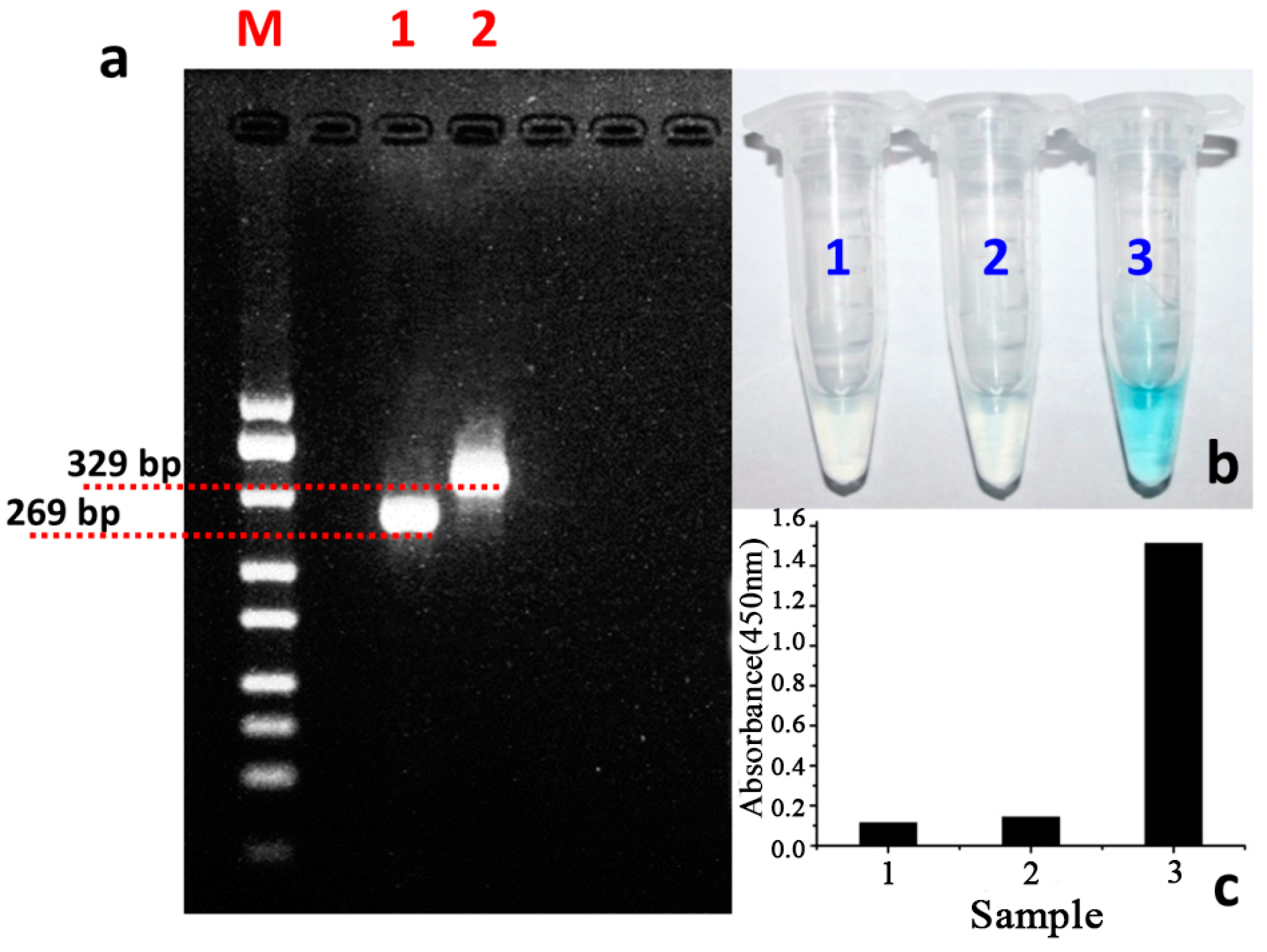
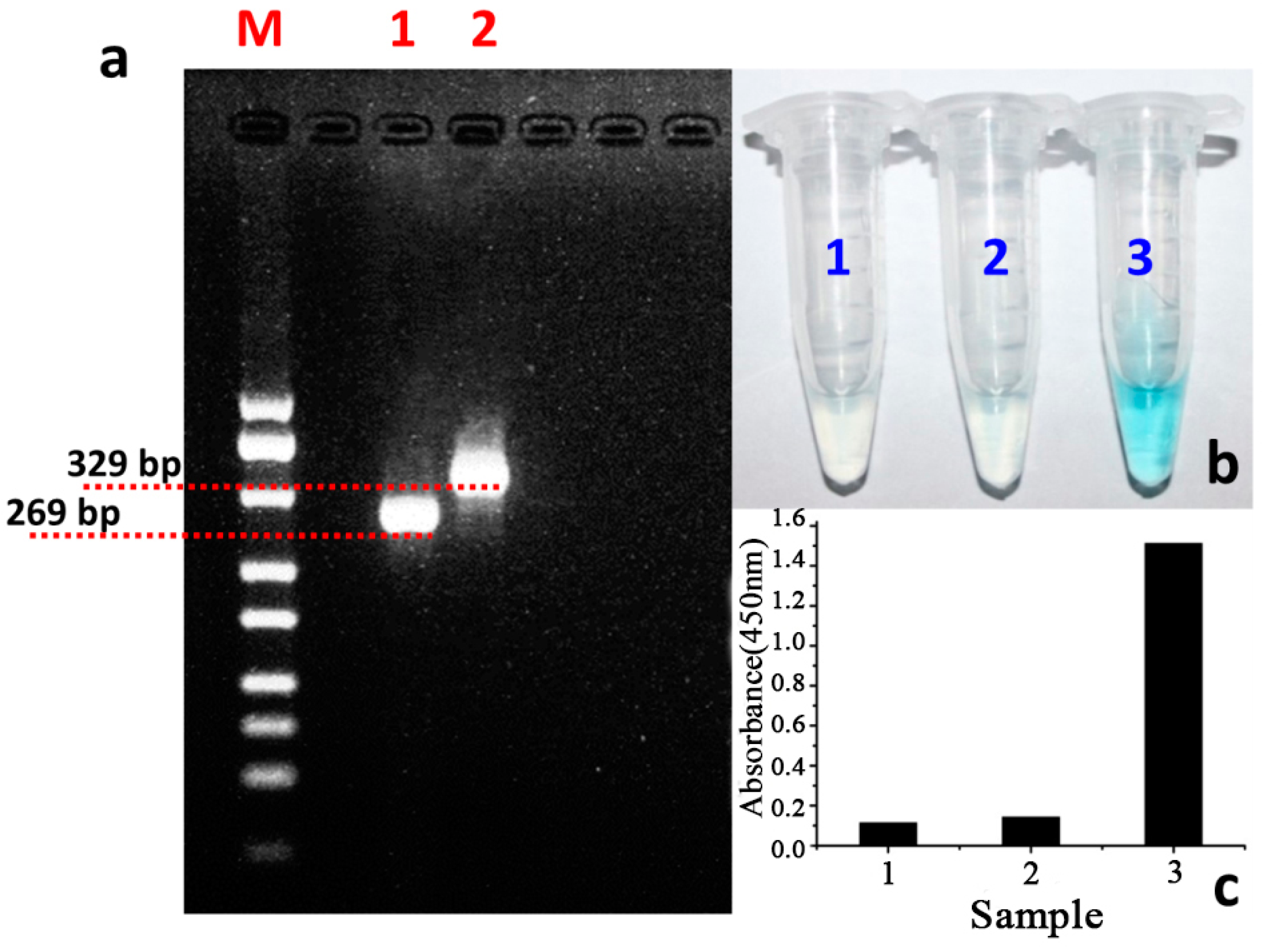
3.2. The Feasibility Study of the Designed Colorimetric Method
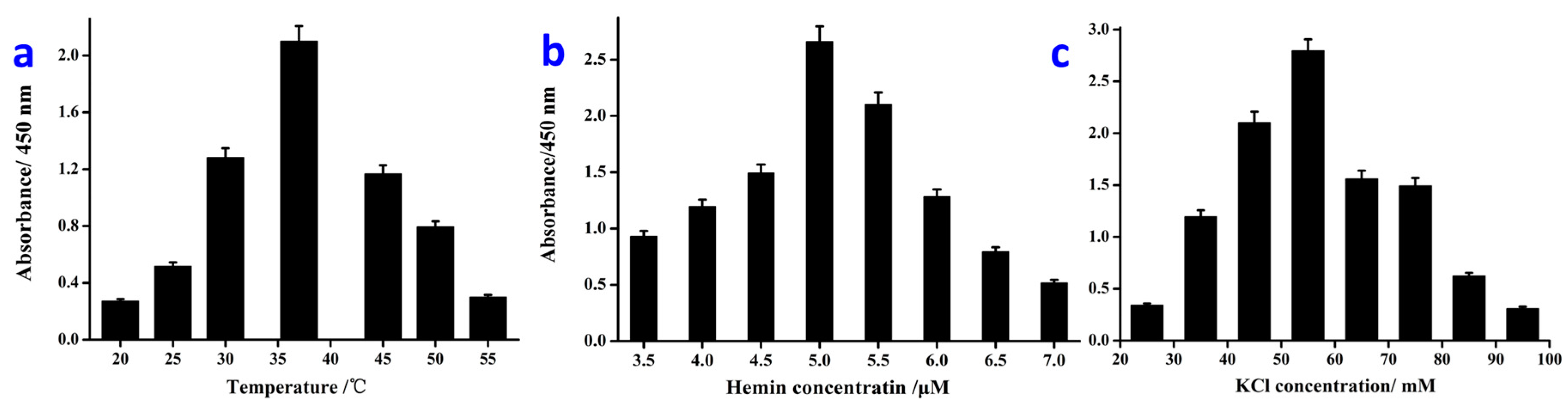
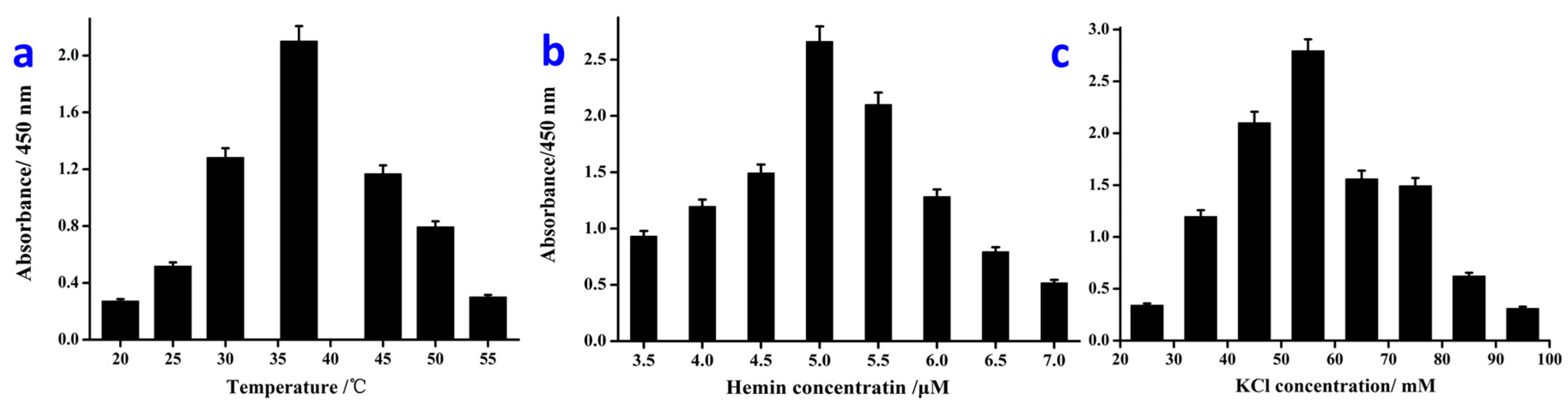
3.3. Optimization of the Detection Conditions
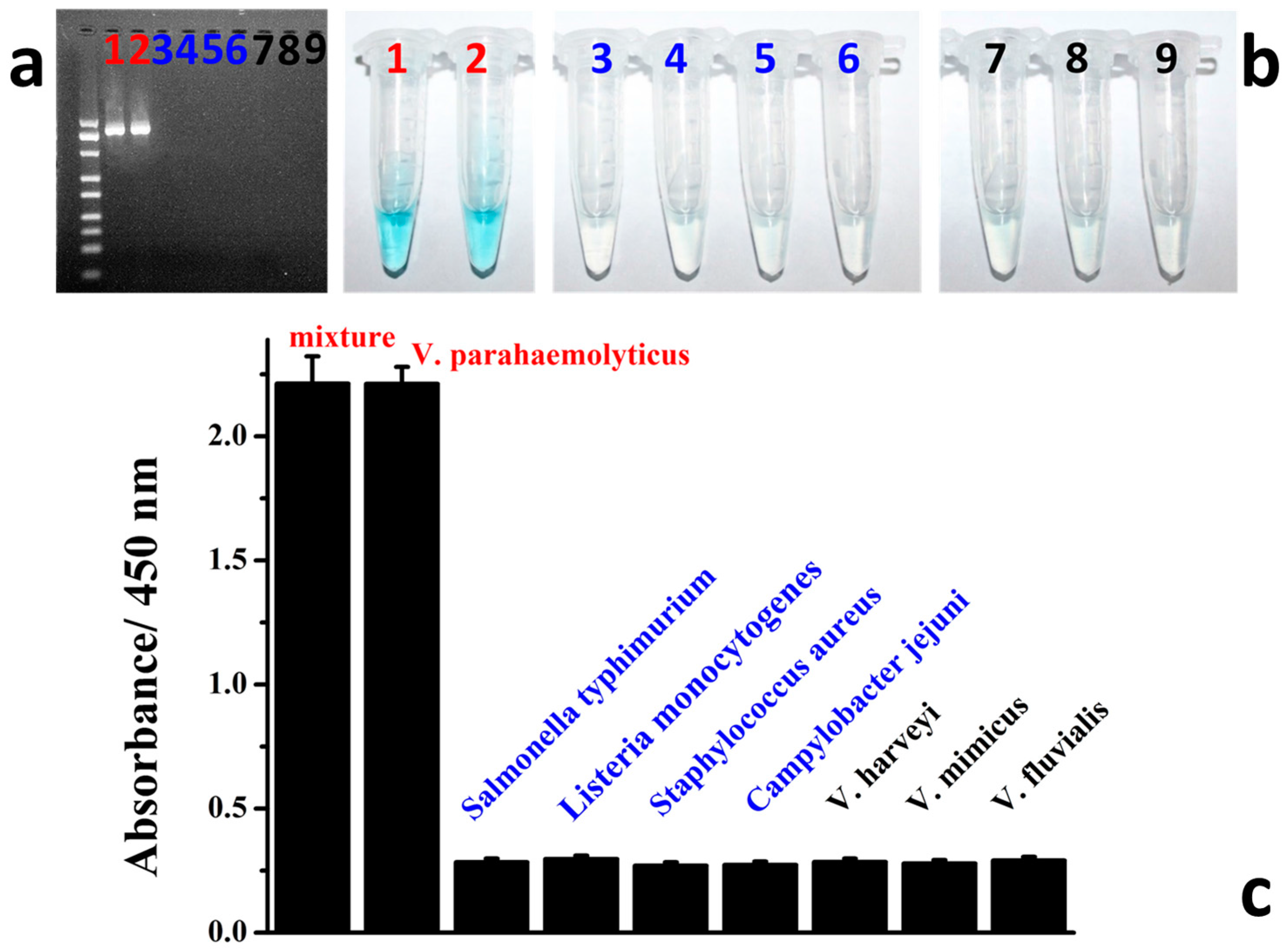
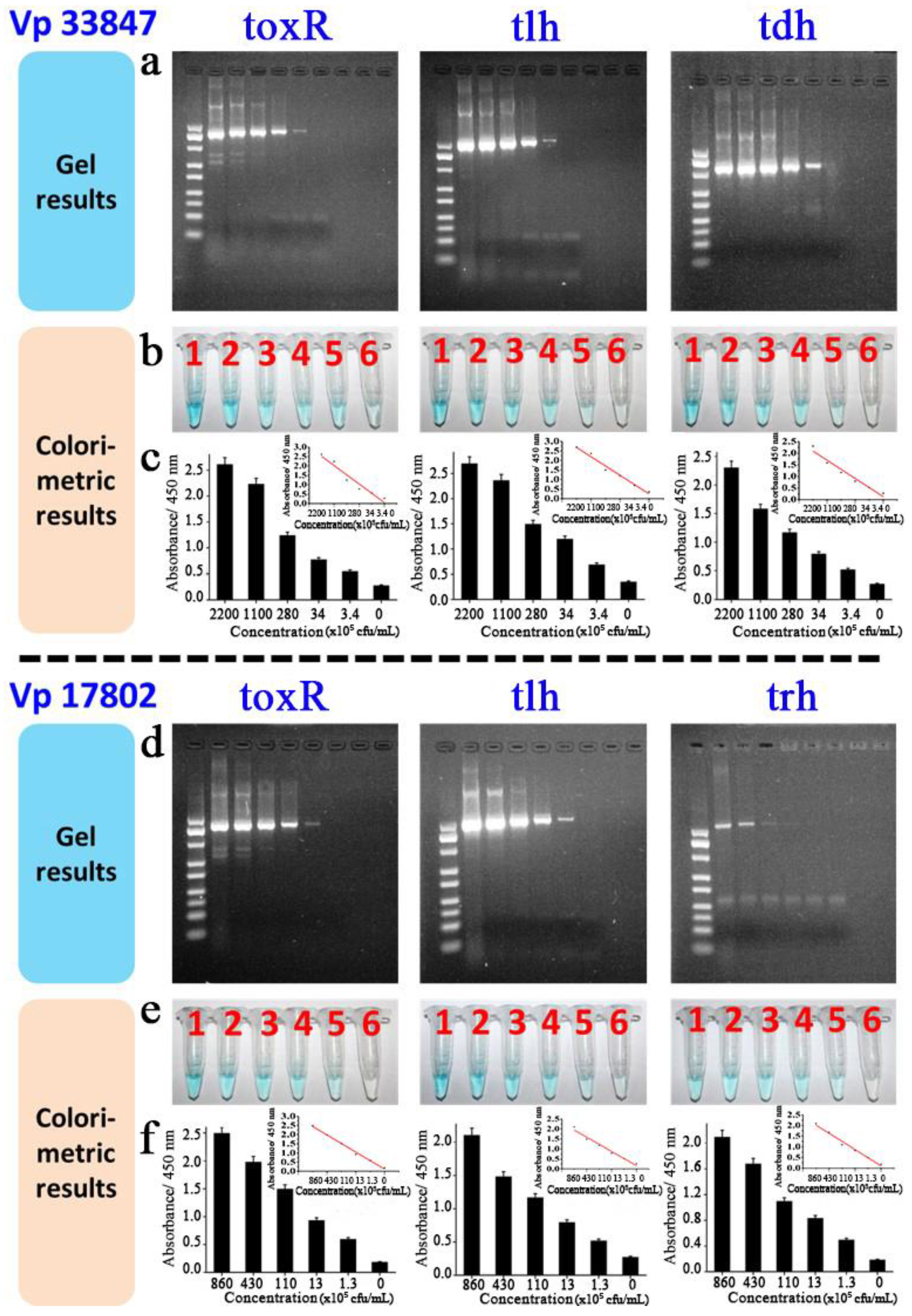
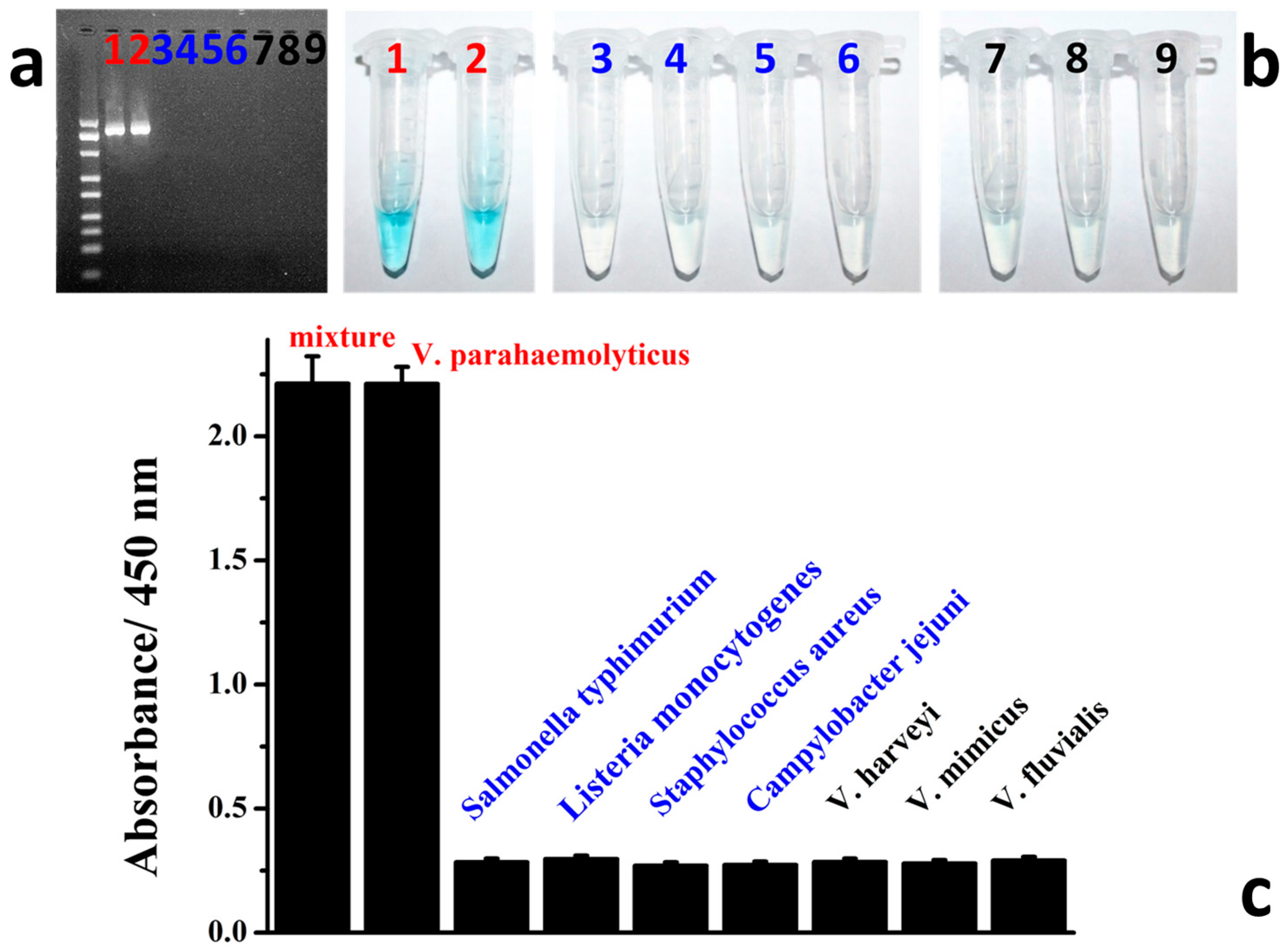
3.4. Colorimetric Detection of Target V. parahaemolyticus
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Zhang, Q.; Dong, X.; Chen, B.; Zhang, Y.; Zu, Y.; Li, W. Zebrafish as a useful model for zoonotic Vibrio parahaemolyticus pathogenicity in fish and human. Dev. Comp. Immunol. 2015, 55, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Hossain, M.T.; Kim, Y.O.; Kong, I.S. Multiplex PCR for the detection and differentiation of Vibrio parahaemolyticus strains using the groEL, tdh and trh genes. Mol. Cell. Probes 2013, 27, 171–175. [Google Scholar] [CrossRef] [PubMed]
- Huehn, S.; Eichhorn, C.; Urmersbach, S.; Breidenbach, J.; Bechlars, S.; Bier, N. Pathogenic vibrios in environmental, seafood and clinical sources in Germany. Int. J. Med. Microbiol. 2014, 304, 843–850. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Wen, J.; Ma, Y.; Ma, X.; Chen, Y. Epidemiology of foodborne disease outbreaks caused by Vibrio parahaemolyticus, China, 2003–2008. Food Control 2014, 46, 197–202. [Google Scholar] [CrossRef]
- Kadhim, H.M.; Miah, A.; Munn, C.B.; Gilpin, M.L. Development of a polymerase chain reaction (PCR) test for the detection of virulent forms of Vibrio parahaemolyticus. Eur. J. Clin. Microbiol. Infect. Dis. 2012, 31, 431–439. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, A.V.; Bej, A.K. Multiplexed real-time PCR amplification of tlh, tdh and trh genes in Vibrio parahaemolyticus and its rapid detection in shellfish and Gulf of Mexico water. Antonie Van Leeuwenhoek 2010, 98, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Yáñez, R.; Bastías, R.; Higuera, G.; Salgado, O.; Katharios, P.; Romero, J. Amplification of tlh gene in other Vibrionaceae specie by specie-specific multiplex PCR of Vibrio parahaemolyticus. Electron. J. Biotechnol. 2015, 18, 459–463. [Google Scholar] [CrossRef]
- He, P.; Chen, Z.; Luo, J.; Wang, H.; Yan, Y.; Chen, L. Multiplex real-time PCR assay for detection of pathogenic Vibrio parahaemolyticus strains. Mol. Cell. Probes 2014, 28, 246–250. [Google Scholar] [CrossRef] [PubMed]
- Parveen, S.; DaSilva, L.; DePaola, A.; Bowers, J.; White, C.; Munasinghe, K.A.; Brohawn, K.; Mudoh, M.; Tamplin, M. Development and validation of a predictive model for the growth of Vibrio parahaemolyticus in post-harvest shellstock oysters. Int. J. Food Microbiol. 2013, 161, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.; Zhao, H.; Xian, Y.; Hussain, M.A.; Wu, X. Multiplex PCR assays for the detection of Vibrio alginolyticus, Vibrio parahaemolyticus, Vibrio vulnificus, and Vibrio cholerae with an internal amplification control. Diagn. Microbiol. Infect. Dis. 2014, 79, 115–118. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Qiao, J.; Li, N.; Qi, L.; Zhang, S. Fluorescent probe for turn-on sensing of l-cysteine by ensemble of AuNCs and polymer protected AuNPs. Anal. Chim. Acta 2015, 879, 97–103. [Google Scholar] [CrossRef] [PubMed]
- He, X.P.; Zang, Y.; James, T.D.; Li, J.; Chen, G.R. Probing disease-related proteins with fluorogenic composite materials. Chem. Soc. Rev. 2014, 44, 4239–4248. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Tran, N.H.; Kim, J.; Yoo, S.Y.; Chung, H. Toehold-mediated DNA displacement-based surface-enhanced Raman scattering DNA sensor utilizing an Au–Ag bimetallic nanodendrite substrate. Anal. Chim. Acta 2015, 885, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Mao, K.; Liu, Y.; Xiao, H.; Chen, Y.; Wu, Z.; Zhou, X. A novel platform for detection of protooncogene based on Au nanocluster enhanced fluorescence. Anal. Methods 2014, 7, 40–44. [Google Scholar] [CrossRef]
- Raghunath, P.; Karunasagar, I.; Karunasagar, I. Improved isolation and detection of pathogenic Vibrio parahaemolyticus from seafood using a new enrichment broth. Int. J. Food Microbiol. 2009, 129, 200–203. [Google Scholar] [CrossRef] [PubMed]
- Khouadja, S.; Suffredini, E.; Baccouche, B.; Croci, L.; Bakhrouf, A. Occurrence of virulence genes among Vibrio cholerae and Vibrio parahaemolyticus strains from treated wastewaters. Environ. Monit. Assess. 2014, 186, 6935–6945. [Google Scholar] [CrossRef] [PubMed]
- Duan, N.; Wu, S.; Chen, X.; Huang, Y.; Wang, Z. Selection and identification of a DNA aptamer targeted to Vibrio parahemolyticus. J. Agric. Food Chem. 2012, 60, 4034–4038. [Google Scholar] [CrossRef] [PubMed]
- Duan, N.; Wu, S.; Ma, X.; Xia, Y.; Wang, Z. A universal fluorescent aptasensor based on AccuBlue dye for the detection of pathogenic bacteria. Anal. Biochem. 2014, 454, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Duan, N.; Shi, Z.; Fang, C.; Wang, Z. Simultaneous aptasensor for multiplex pathogenic bacteria detection based on multicolor upconversion nanoparticles labels. Anal. Chem. 2014, 86, 3100–3107. [Google Scholar] [CrossRef] [PubMed]
- Duan, N.; Wu, S.; Zhu, C.; Ma, X.; Wang, Z.; Yu, Y. Dual-color upconversion fluorescence and aptamer-functionalized magnetic nanoparticles-based bioassay for the simultaneous detection of Salmonella Typhimurium and Staphylococcus aureus. Anal. Chim. Acta 2012, 723, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Lu, C.H.; Liu, X.; Freage, L.; Willner, I. Amplified and multiplexed detection of DNA using the dendritic rolling circle amplified synthesis of DNAzyme reporter units. Anal. Chem. 2014, 86, 1614–1621. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yang, Y.; Li, L.; Sun, M.; Yin, H.; Qin, W. A polymeric liquid membrane electrode responsive to 3,3′,5,5′-tetramethylbenzidine oxidation for sensitive peroxidase/peroxidase mimetic-based potentiometric biosensing. Anal. Chem. 2014, 86, 4416–4422. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Kan, Y.Y.; Jiang, J.H.; Yu, R.Q. A simple and highly sensitive DNAzyme-based assay for nicotinamide adenine dinucleotide by ligase-mediated inhibition of strand displacement amplification. Anal. Chim. Acta 2014, 844, 70–74. [Google Scholar] [CrossRef] [PubMed]
- Mun, H.; Jo, E.J.; Li, T.; Joung, H.A.; Hong, D.G.; Shim, W.B. Homogeneous assay of target molecules based on chemiluminescence resonance energy transfer (CRET) using DNAzyme-linked aptamers. Biosens. Bioelectron. 2014, 58, 308–313. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Zhang, Y.; Zhang, Y.; Li, H.; Yang, H.; Wei, H. Sensitive and rapid detection of staphylococcus aureus in milk via cell binding domain of lysin. Biosens. Bioelectron. 2016, 77, 366–371. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.W.; Sun, C.J.; Zhang, F.T.; Xu, L.; Zhou, Y.L.; Zhang, X.X. An electrochemical aptasensor based on enzyme linked aptamer assay. Biosens. Bioelectron. 2012, 31, 363–368. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Wang, Z.; Hu, D.; Ma, Q.; Huang, L.; Xv, C. Scanometric nanomolar lead (II) detection using DNA-functionalized gold nanoparticles and silver stain enhancement. Sens. Actuators B Chem. 2014, 200, 310–316. [Google Scholar] [CrossRef]
- Wu, S.; Wang, Y.; Duan, N.; Ma, H.; Wang, Z. Colorimetric Aptasensor Based on Enzyme for the Detection of Vibrio parahemolyticus. J. Agric. Food Chem. 2015, 63, 7849–7854. [Google Scholar] [CrossRef] [PubMed]
- Ginnari, M.; Ghidini, V.; Caburlotto, G.; Lleo, M.M. Virulence genes and pathogenicity islands in environmental Vibrio strains nonpathogenic to humans. FEMS Microbiol. Ecol. 2012, 82, 563–573. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, K.; Pan, D.; Teng, J.; Yao, L.; Ye, Y.; Xue, F.; Xia, F.; Chen, W. Colorimetric Integrated PCR Protocol for Rapid Detection of Vibrio parahaemolyticus. Sensors 2016, 16, 1600. https://doi.org/10.3390/s16101600
Cheng K, Pan D, Teng J, Yao L, Ye Y, Xue F, Xia F, Chen W. Colorimetric Integrated PCR Protocol for Rapid Detection of Vibrio parahaemolyticus. Sensors. 2016; 16(10):1600. https://doi.org/10.3390/s16101600
Chicago/Turabian StyleCheng, Kewen, Daodong Pan, Jun Teng, Li Yao, Yingwang Ye, Feng Xue, Fan Xia, and Wei Chen. 2016. "Colorimetric Integrated PCR Protocol for Rapid Detection of Vibrio parahaemolyticus" Sensors 16, no. 10: 1600. https://doi.org/10.3390/s16101600
APA StyleCheng, K., Pan, D., Teng, J., Yao, L., Ye, Y., Xue, F., Xia, F., & Chen, W. (2016). Colorimetric Integrated PCR Protocol for Rapid Detection of Vibrio parahaemolyticus. Sensors, 16(10), 1600. https://doi.org/10.3390/s16101600