
Continuous-Wave Stimulated Emission Depletion Microscope for Imaging Actin Cytoskeleton in Fixed and Live Cells


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Experimental Section
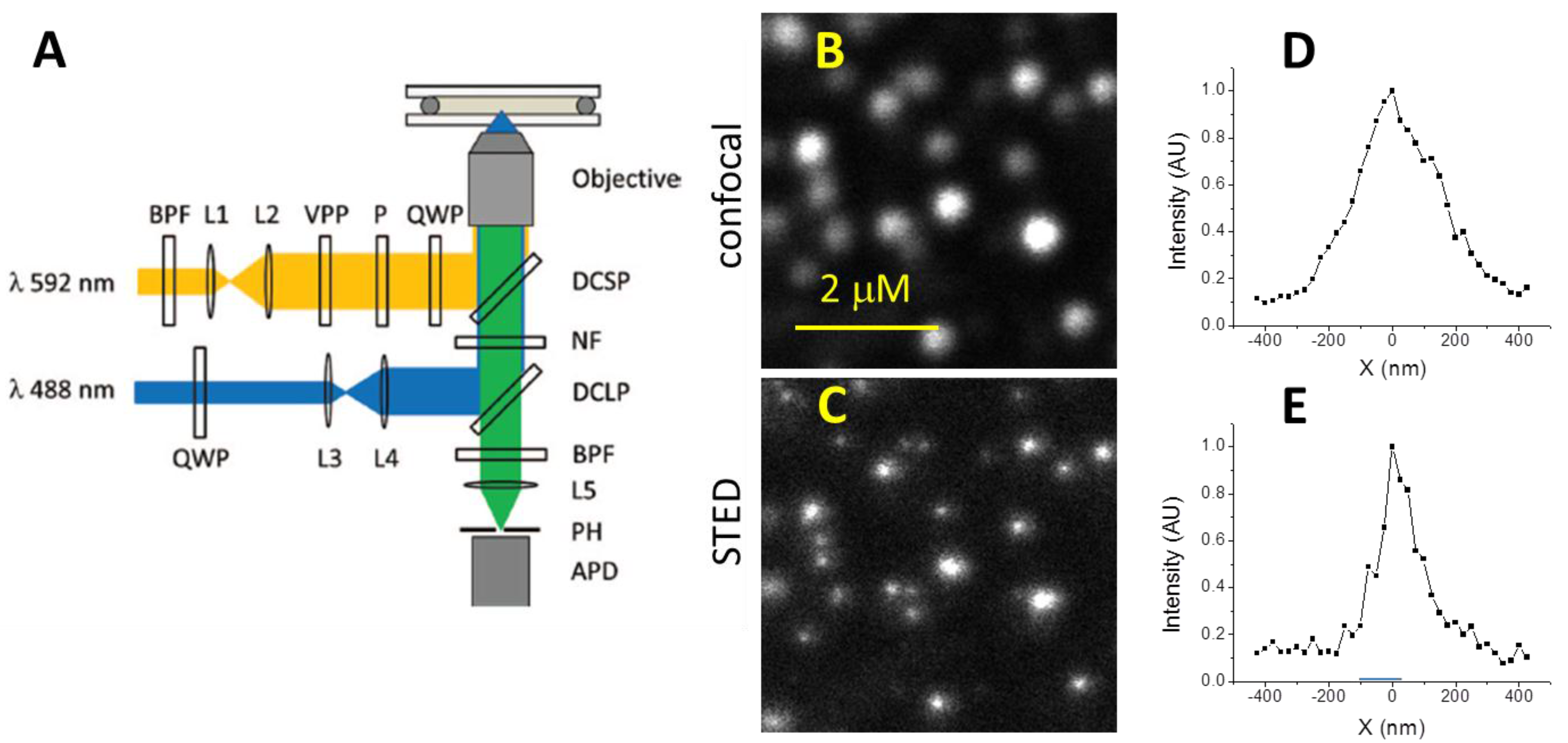
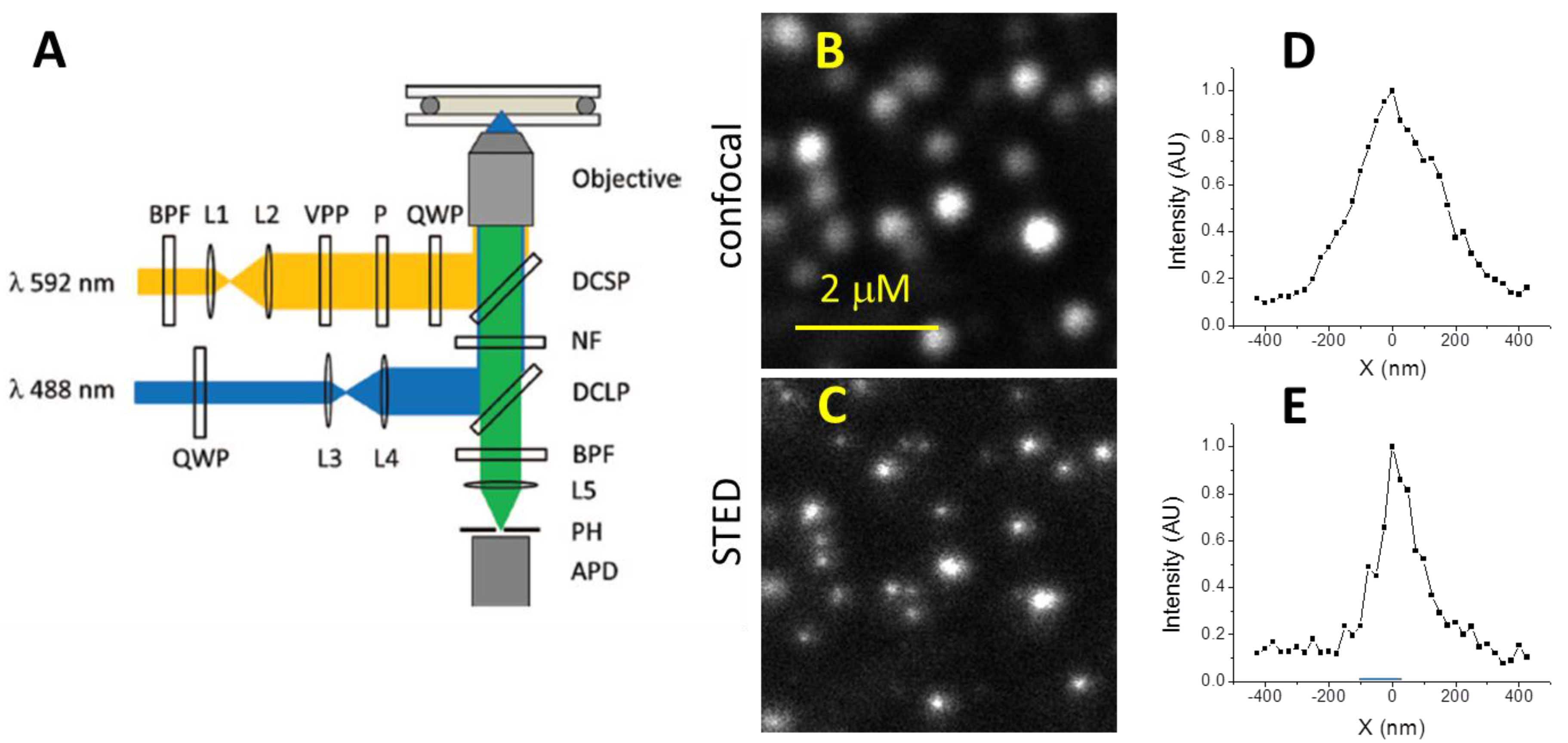
2.1. STED Microscope Setup
2.2. Cell Culture and Labeling of PC-12 Cells
2.3. Cell Culture and Transfection of RCS Cells
3. Results and Discussion
3.1. Resolution of STED Microscopy
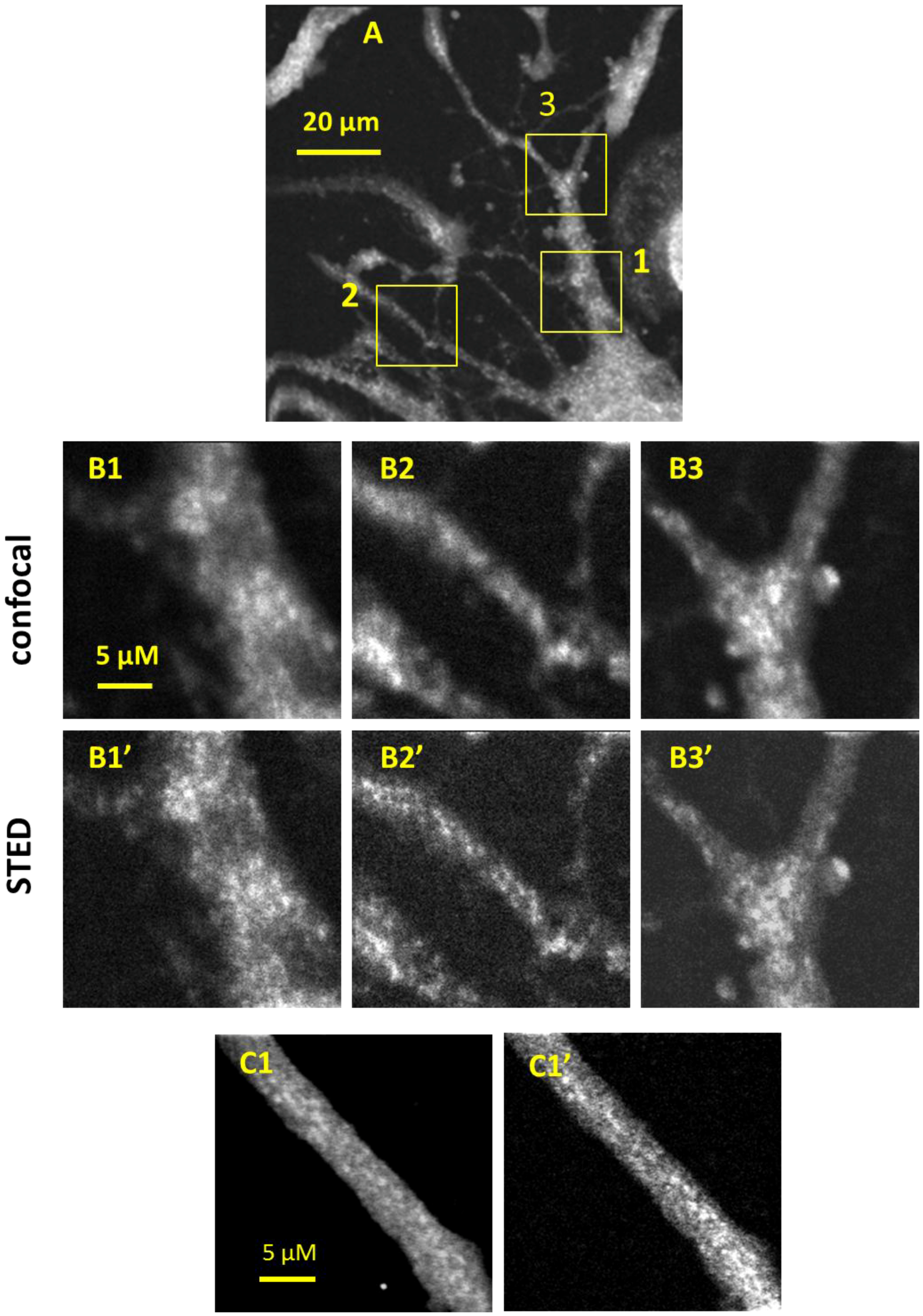
3.2. CW-STED Imaging of Fixed PC-12 Cells
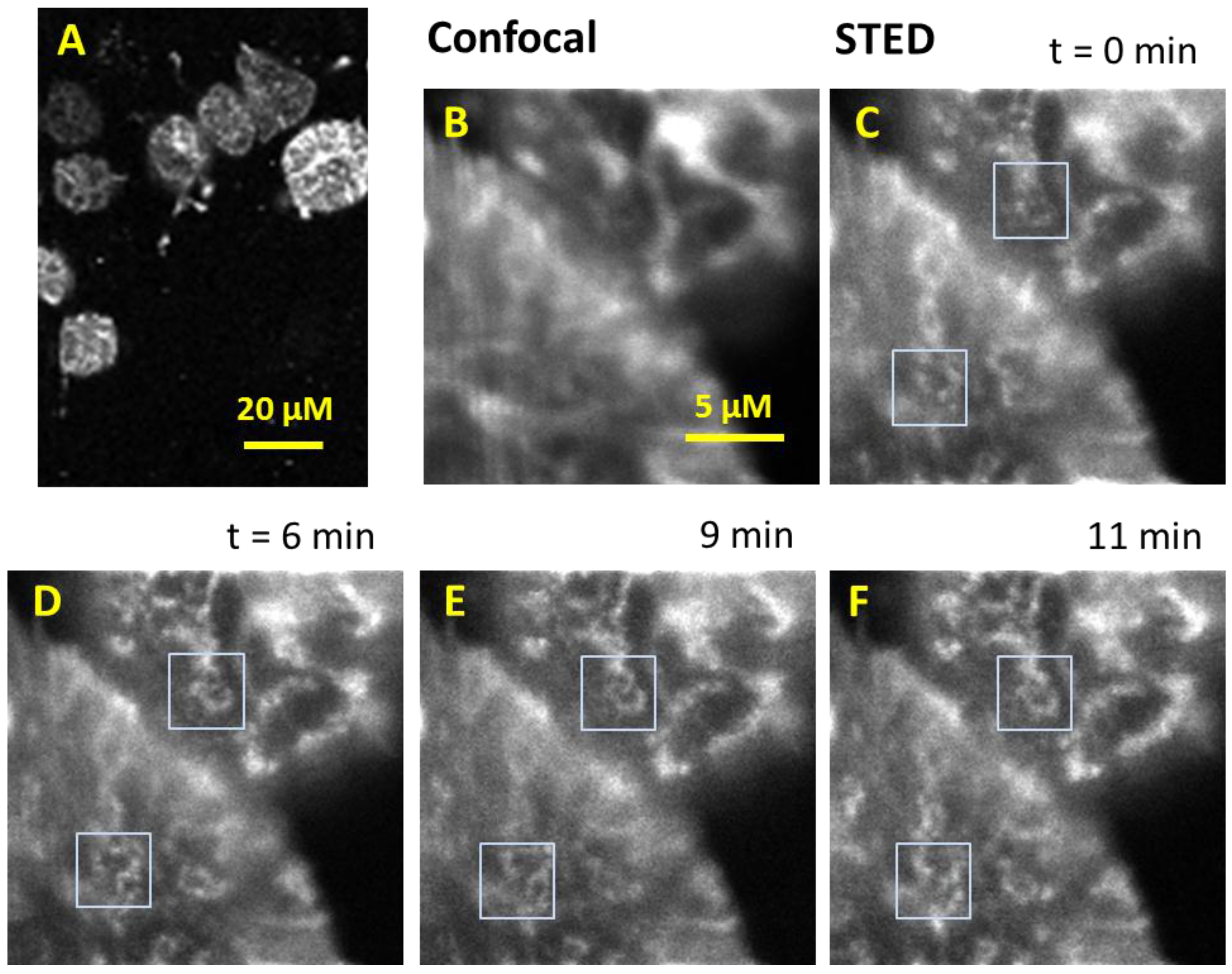
3.3. CW-STED Imaging of Live Chondrocytes
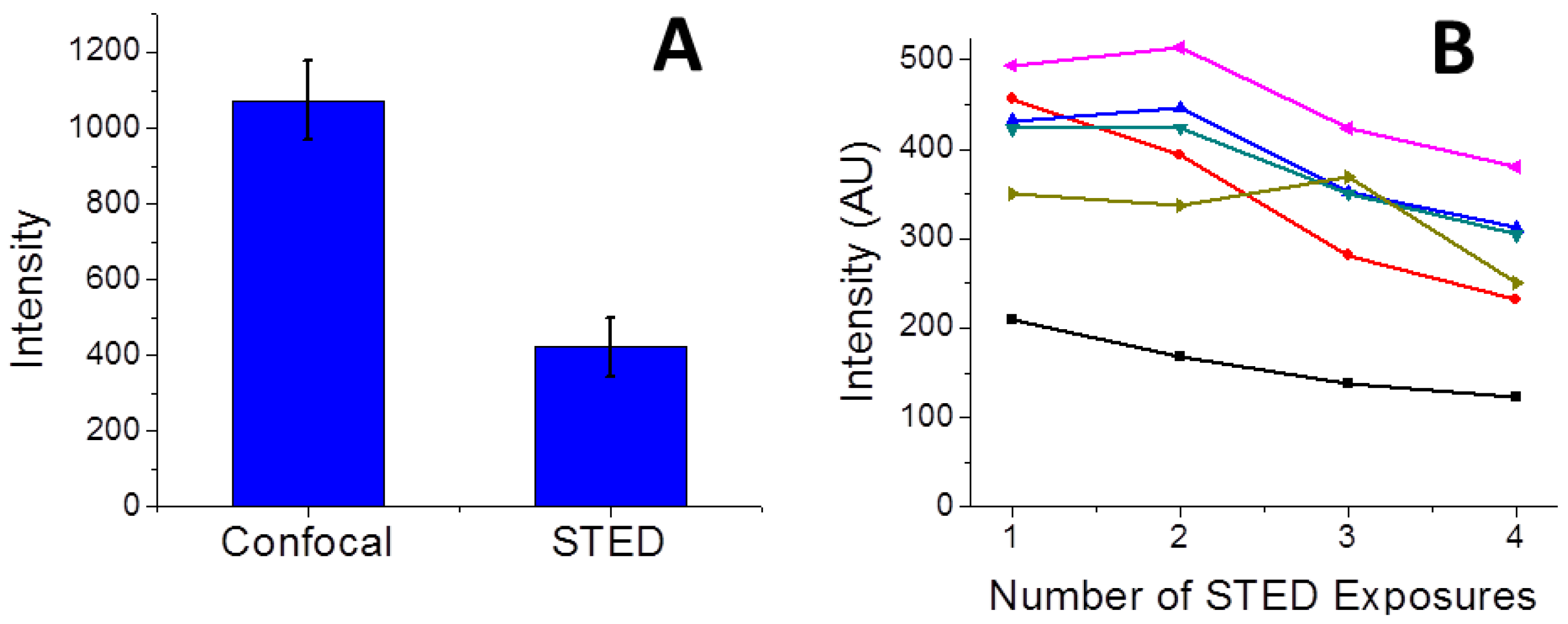
3.4. Challenges and Opportunities
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Mortensen, K.I.; Churchman, L.S.; Spudich, J.A.; Flyvbjerg, H. Optimized localization analysis for single-molecule tracking and super-resolution microscopy. Nat. Methods 2010, 7, U377–U359. [Google Scholar] [CrossRef] [PubMed]
- Baddeley, D.; Crossman, D.; Rossberger, S.; Cheyne, J.E.; Montgomery, J.M.; Jayasinghe, I.D.; Cremer, C.; Cannell, M.B.; Soeller, C. 4D super-resolution microscopy with conventional fluorophores and single wavelength excitation in optically thick cells and tissues. PLoS ONE 2011, 6. [Google Scholar] [CrossRef] [PubMed]
- Zanacchi, F.C.; Lavagnino, Z.; Donnorso, M.P.; del Bue, A.; Furia, L.; Faretta, M.; Diaspro, A. Live-cell 3D super-resolution imaging in thick biological samples. Nat. Methods 2011, 8, 1047–1049. [Google Scholar] [CrossRef] [PubMed]
- Eggeling, C.; Willig, K.I.; Barrantes, F.J. Sted microscopy of living cells-new frontiers in membrane and neurobiology. J. Neurochem. 2013, 126, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Betzig, E.; Trautman, J.K. Near-field optics-microscopy, spectroscopy, and surface modification beyond the diffraction limit. Science 1992, 257, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Hell, S.W.; Wichmann, J. Breaking the diffraction resolution limit by stimulated-emission-stimulated-emission-depletion fluorescence microscopy. Opt. Lett. 1994, 19, 780–782. [Google Scholar] [CrossRef] [PubMed]
- Klar, T.A.; Jakobs, S.; Dyba, M.; Egner, A.; Hell, S.W. Fluorescence microscopy with diffraction resolution barrier broken by stimulated emission. Proc. Natl. Acad. Sci. U.S.A. 2000, 97, 8206–8210. [Google Scholar] [CrossRef] [PubMed]
- Rust, M.J.; Bates, M.; Zhuang, X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (storm). Nat. Methods 2006, 3, 793–795. [Google Scholar] [CrossRef] [PubMed]
- Betzig, E.; Patterson, G.H.; Sougrat, R.; Lindwasser, O.W.; Olenych, S.; Bonifacino, J.S.; Davidson, M.W.; Lippincott-Schwartz, J.; Hess, H.F. Imaging intracellular fluorescent proteins at nanometer resolution. Science 2006, 313, 1642–1645. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Wang, W.; Bates, M.; Zhuang, X. Three-dimensional super-resolution imaging by stochastic optical reconstruction microscopy. Science 2008, 319, 810–813. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, M.; Eggeling, C.; Jakobs, S.; Hell, S.W. Breaking the diffraction barrier in fluorescence microscopy at low light intensities by using reversibly photoswitchable proteins. Proc. Natl. Acad. Sci. 2005, 102, 17565–17569. [Google Scholar] [CrossRef] [PubMed]
- Hell, S.W. Far-field optical nanoscopy. Science 2007, 316, 1153–1158. [Google Scholar] [CrossRef] [PubMed]
- Gustafsson, M.G.L.; Agard, D.A.; Sedat, J.W. I5M: 3D widefield light microscopy with better than 100 nm axial resolution. J. Microsc. 1999, 195, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhao, M.; Peng, L. Nonlinear structured illumination microscopy by surface plasmon enhanced stimulated emission depletion. Opt. Express 2011, 19, 24783–24794. [Google Scholar] [CrossRef] [PubMed]
- Gustafsson, M.G.L. Surpassing the lateral resolution limit by a factor of two using structured illumination microscopy. J. Microsc. 2000, 198, 82–87. [Google Scholar] [CrossRef] [PubMed]
- Gustafsson, M.G.L. Nonlinear structured-illumination microscopy: Wide-field fluorescence imaging with theoretically unlimited resolution. Proc. Natl. Acad. Sci. 2005, 102, 13081–13086. [Google Scholar] [CrossRef] [PubMed]
- Rittweger, E.; Han, K.Y.; Irvine, S.E.; Eggeling, C.; Hell, S.W. Sted microscopy reveals crystal colour centres with nanometric resolution. Nat. Photon. 2009, 3, 144–147. [Google Scholar] [CrossRef]
- Westphal, V.; Rizzoli, S.O.; Lauterbach, M.A.; Kamin, D.; Jahn, R.; Hell, S.W. Video-rate far-field optical nanoscopy dissects synaptic vesicle movement. Science 2008, 320, 246–249. [Google Scholar] [CrossRef] [PubMed]
- Chmyrov, A.; Keller, J.; Grotjohann, T.; Ratz, M.; d’Este, E.; Jakobs, S.; Eggeling, C.; Hell, S.W. Nanoscopy with more than 100000 “doughnuts”. Nat. Methods 2013, 10, 737–740. [Google Scholar] [CrossRef] [PubMed]
- Moneron, G.; Medda, R.; Hein, B.; Giske, A.; Westphal, V.; Hell, S.W. Fast sted microscopy with continuous wave fiber lasers. Opt. Express 2010, 18, 1302–1309. [Google Scholar] [CrossRef] [PubMed]
- Neupane, B.; Ligler, F.S.; Wang, G. Review of recent developments in stimulated emission depletion microscopy: Applications on cell imaging. J. Biomed. Opt. 2014, 19. [Google Scholar] [CrossRef] [PubMed]
- Neupane, B.; Chen, F.; Sun, W.; Chiu, D.T.; Wang, G. Tuning donut profile for spatial resolution in stimulated emission depletion microscopy. Rev. Sci. Instrum. 2013, 84. [Google Scholar] [CrossRef] [PubMed]
- Clarke, M.; Spudich, J.A. Non-muscle contractile proteins-role of actin and myosin in cell motility and shape determination. Annu. Rev. Biochem. 1977, 46, 797–822. [Google Scholar] [CrossRef] [PubMed]
- Pollard, T.D.; Cooper, J.A. Actin, a central player in cell shape and movement. Science 2009, 326, 1208–1212. [Google Scholar] [CrossRef] [PubMed]
- Bennett, V. The spectrin-actin junction of erythrocyte-membrane skeletons. Biochim. Et. Biophys. Acta 1989, 988, 107–121. [Google Scholar] [CrossRef]
- Mitchison, T.J.; Cramer, L.P. Actin-based cell motility and cell locomotion. Cell 1996, 84, 371–379. [Google Scholar] [CrossRef]
- Pardee, J.D.; Spudich, J.A. Purification of muscle actin. Methods Enzymol. 1982, 85, 164–181. [Google Scholar] [PubMed]
- Hall, A. Rho gtpases and the actin cytoskeleton. Science 1998, 279, 509–514. [Google Scholar] [CrossRef] [PubMed]
- Stossel, T.P. From signal to pseudopod—how cells control cytoplasmic actin assembly. J. Biol. Chem. 1989, 264, 18261–18264. [Google Scholar] [PubMed]
- Ridley, A.J. Rho gtpases and actin dynamics in membrane protrusions and vesicle trafficking. Trends Cell Biol. 2006, 16, 522–529. [Google Scholar] [CrossRef] [PubMed]
- Pollard, T.D.; Weihing, R.R. Actin and myosin and cell movement. Crit. Rev. Biochem. 1974, 2, 1–65. [Google Scholar]
- Doherty, G.J.; McMahon, H.T. Mediation, modulation, and consequences of membrane-cytoskeleton interactions. Ann. Rev. Biophys. 2008, 37, 65–95. [Google Scholar] [CrossRef] [PubMed]
- Tonnesen, J.; Naegerl, U.V. Superresolution imaging for neuroscience. Exp. Neurol. 2013, 242, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Bacskai, B.J.; Wallen, P.; Levram, V.; Grillner, S.; Tsien, R.Y. Activity-related calcium dynamics in lamprey motoneurons as revealed by video-rate confocal microscopy. Neuron 1995, 14, 19–28. [Google Scholar] [CrossRef]
- Han, X.; Qian, X.; Bernstein, J.G.; Zhou, H.-H.; Franzesi, G.T.; Stern, P.; Bronson, R.T.; Graybiel, A.M.; Desimone, R.; Boyden, E.S. Millisecond-timescale optical control of neural dynamics in the nonhuman primate brain. Neuron 2009, 62, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Stender, A.S.; Marchuk, K.; Liu, C.; Sander, S.; Meyer, M.W.; Smith, E.A.; Neupane, B.; Wang, G.F.; Li, J.J.; Cheng, J.X.; et al. Single cell optical imaging and spectroscopy. Chem. Rev. 2013, 113, 2469–2527. [Google Scholar] [PubMed]
- Lukinavicius, G.; Reymond, L.; D’Este, E.; Masharina, A.; Gottfert, F.; Ta, H.; Guther, A.; Fournier, M.; Rizzo, S.; Waldmann, H.; et al. Fluorogenic probes for live-cell imaging of the cytoskeleton. Nat. Methods 2014, 11, 731–733. [Google Scholar] [CrossRef] [PubMed]
- Urban, N.T.; Willig, K.I.; Hell, S.W.; Nagerl, U.V. Sted nanoscopy of actin dynamics in synapses deep inside living brain slices. Biophys. J. 2011, 101, 1277–1284. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Zhong, G.; Zhuang, X. Actin, spectrin, and associated proteins form a periodic cytoskeletal structure in axons. Science 2013, 339, 452–456. [Google Scholar] [CrossRef] [PubMed]
- Bennett, V.; Davis, J.; Fowler, W.E. Brain spectrin, a membrane-associated protein related in structure and function to erythrocyte spectrin. Nature 1982, 299, 126–131. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.U.; Meyer, K.; Swarm, R. Mucopolysaccharide and protein--polysaccharide of a transplantable rat chondrosarcoma. Proc. Natl. Acad. Sci. 1971, 68, 877–879. [Google Scholar] [CrossRef] [PubMed]
- Wieland, T.; Faulstich, H. Amatoxins, phallotoxins, phallolysin, and antamanide-biologically-active components of poisonous amanita mushrooms. CRC Crit. Rev. Biochem. 1978, 5, 185–260. [Google Scholar] [CrossRef] [PubMed]
- Hein, B.; Willig, K.I.; Wurm, C.A.; Westphal, V.; Jakobs, S.; Hell, S.W. Stimulated emission depletion nanoscopy of living cells using snap-tag fusion proteins. Biophys. J. 2010, 98, 158–163. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Neupane, B.; Jin, T.; Mellor, L.F.; Loboa, E.G.; Ligler, F.S.; Wang, G. Continuous-Wave Stimulated Emission Depletion Microscope for Imaging Actin Cytoskeleton in Fixed and Live Cells. Sensors 2015, 15, 24178-24190. https://doi.org/10.3390/s150924178
Neupane B, Jin T, Mellor LF, Loboa EG, Ligler FS, Wang G. Continuous-Wave Stimulated Emission Depletion Microscope for Imaging Actin Cytoskeleton in Fixed and Live Cells. Sensors. 2015; 15(9):24178-24190. https://doi.org/10.3390/s150924178
Chicago/Turabian StyleNeupane, Bhanu, Tao Jin, Liliana F. Mellor, Elizabeth G. Loboa, Frances S. Ligler, and Gufeng Wang. 2015. "Continuous-Wave Stimulated Emission Depletion Microscope for Imaging Actin Cytoskeleton in Fixed and Live Cells" Sensors 15, no. 9: 24178-24190. https://doi.org/10.3390/s150924178
APA StyleNeupane, B., Jin, T., Mellor, L. F., Loboa, E. G., Ligler, F. S., & Wang, G. (2015). Continuous-Wave Stimulated Emission Depletion Microscope for Imaging Actin Cytoskeleton in Fixed and Live Cells. Sensors, 15(9), 24178-24190. https://doi.org/10.3390/s150924178