
Modulation of Intracellular Quantum Dot to Fluorescent Protein Förster Resonance Energy Transfer via Customized Ligands and Spatial Control of Donor–Acceptor Assembly



Abstract
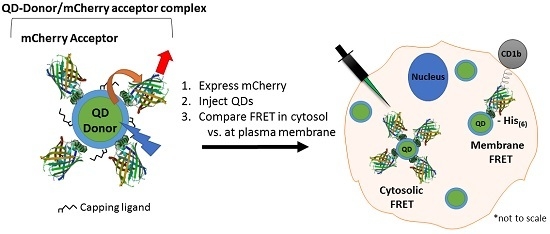
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Experimental
2.1. Materials
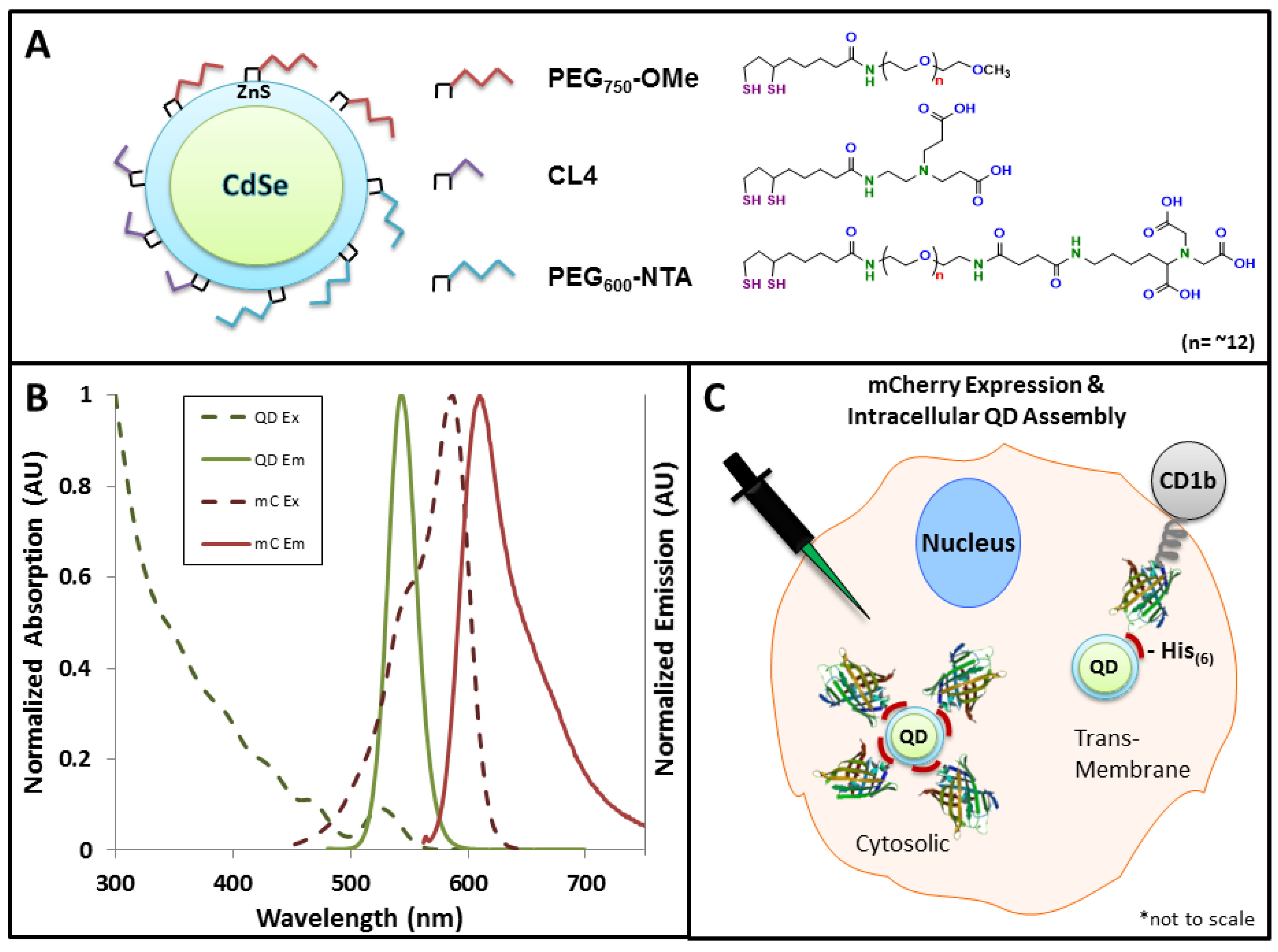
2.2. QD Synthesis and Capping Ligands
2.3. Cell Culture and Transfection
2.4. Microinjection
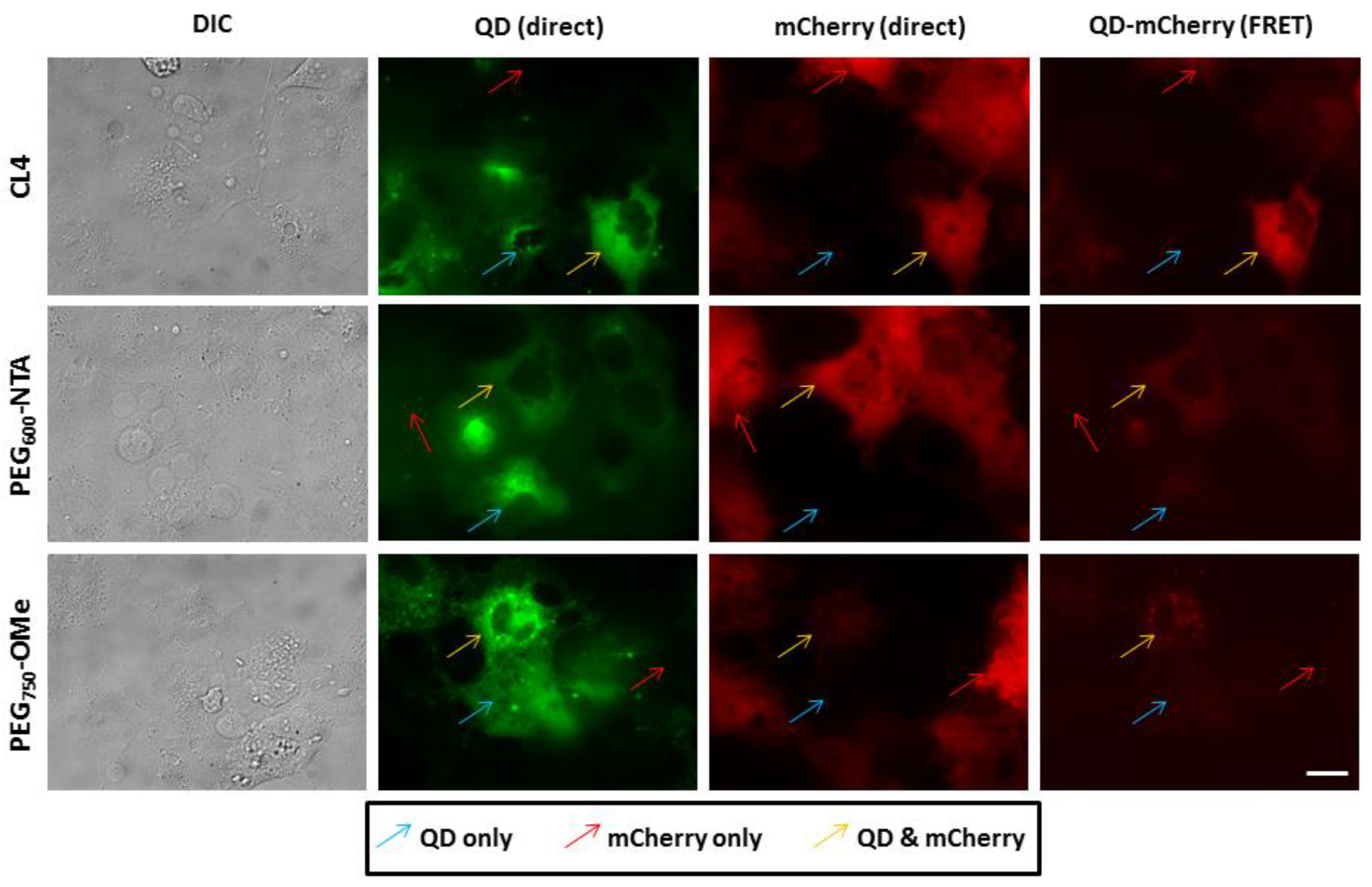
2.5. Microscopy and Image Analysis
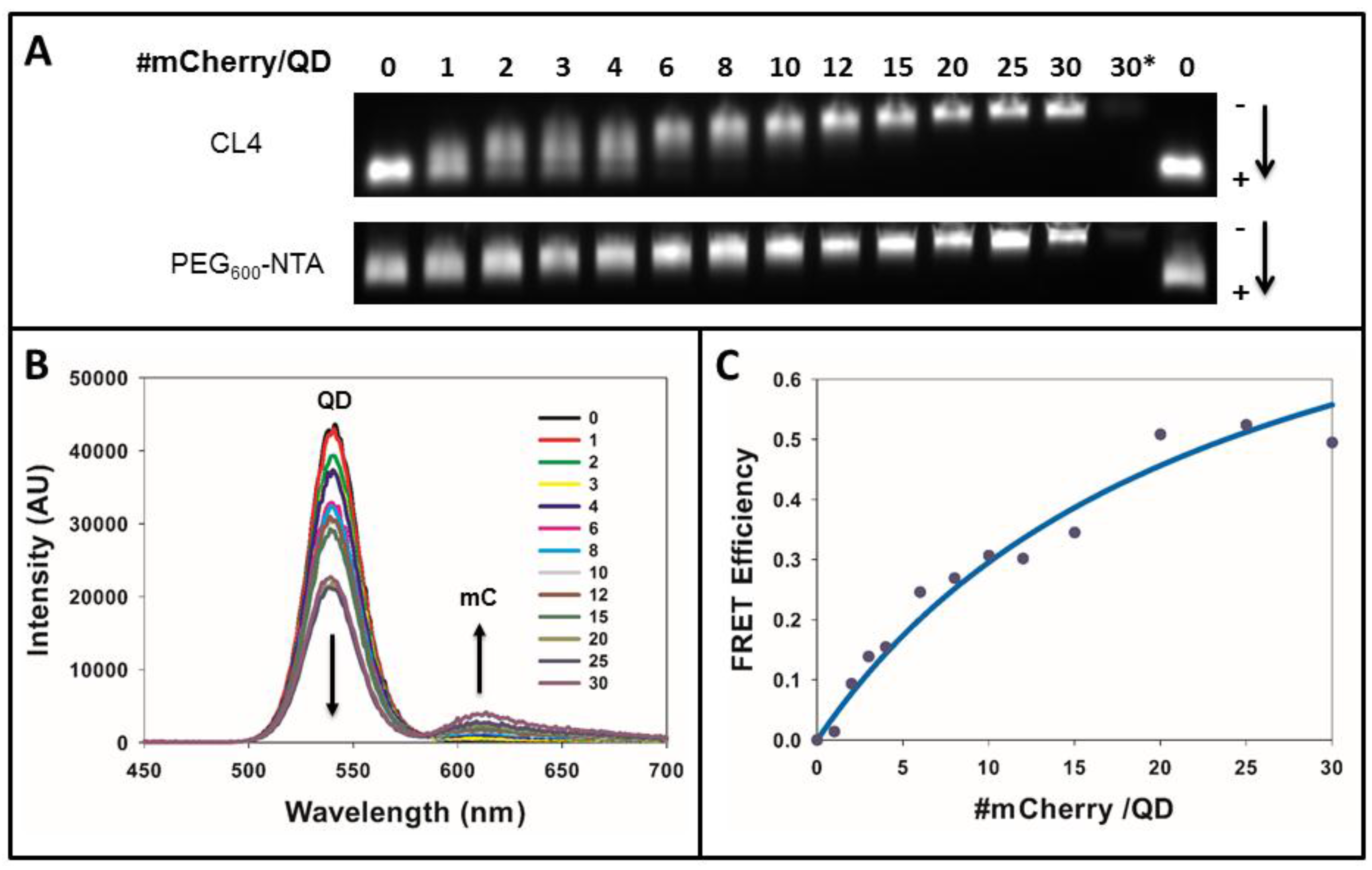
2.6. Gel Electrophoresis
2.7. FRET Analysis
3. Results
3.1. Experimental Rational and Design

3.2. Efficacy of the FRET System

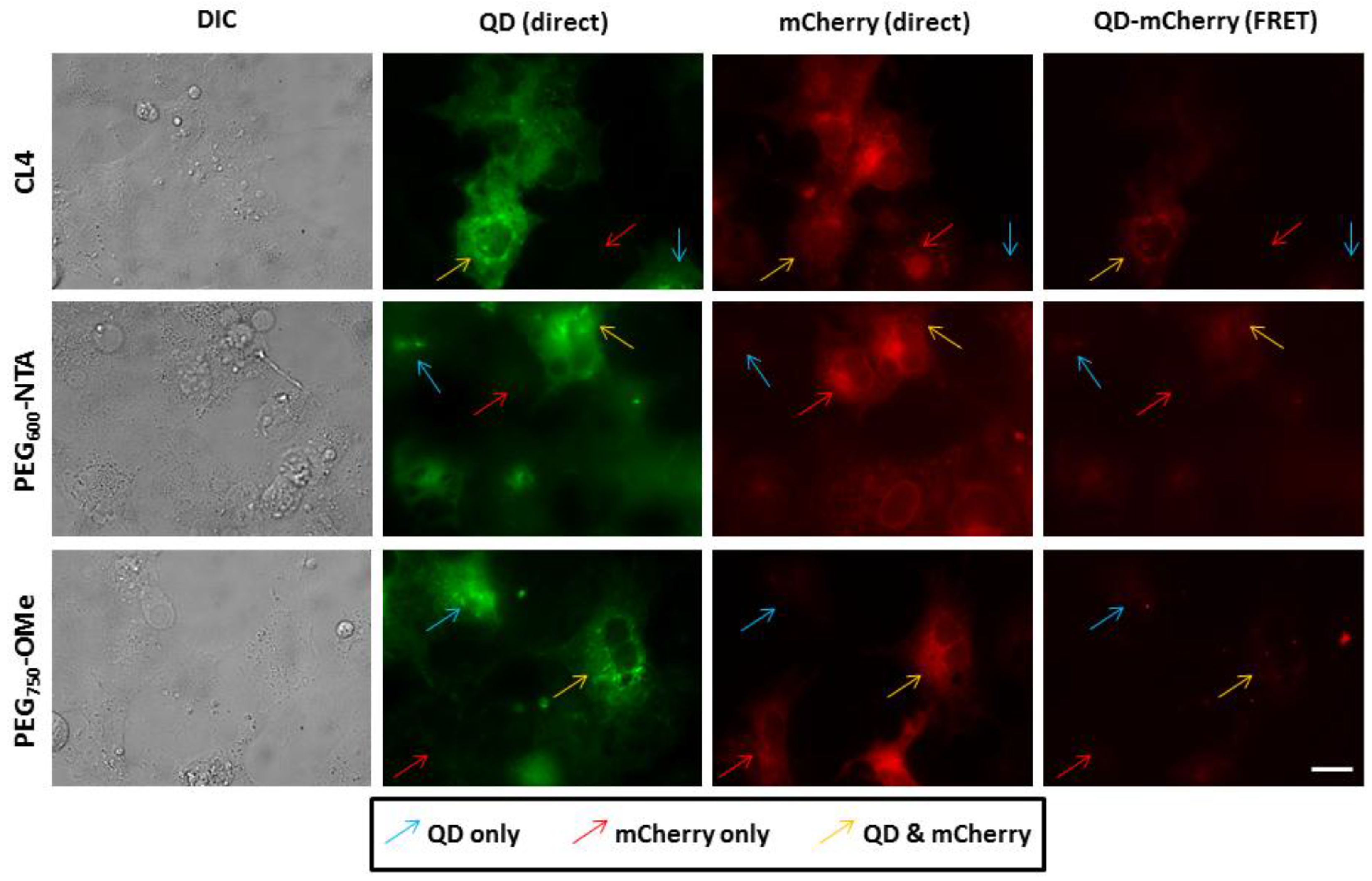
3.3. Intracellular QD-mCherry Assembly and FRET Efficiency
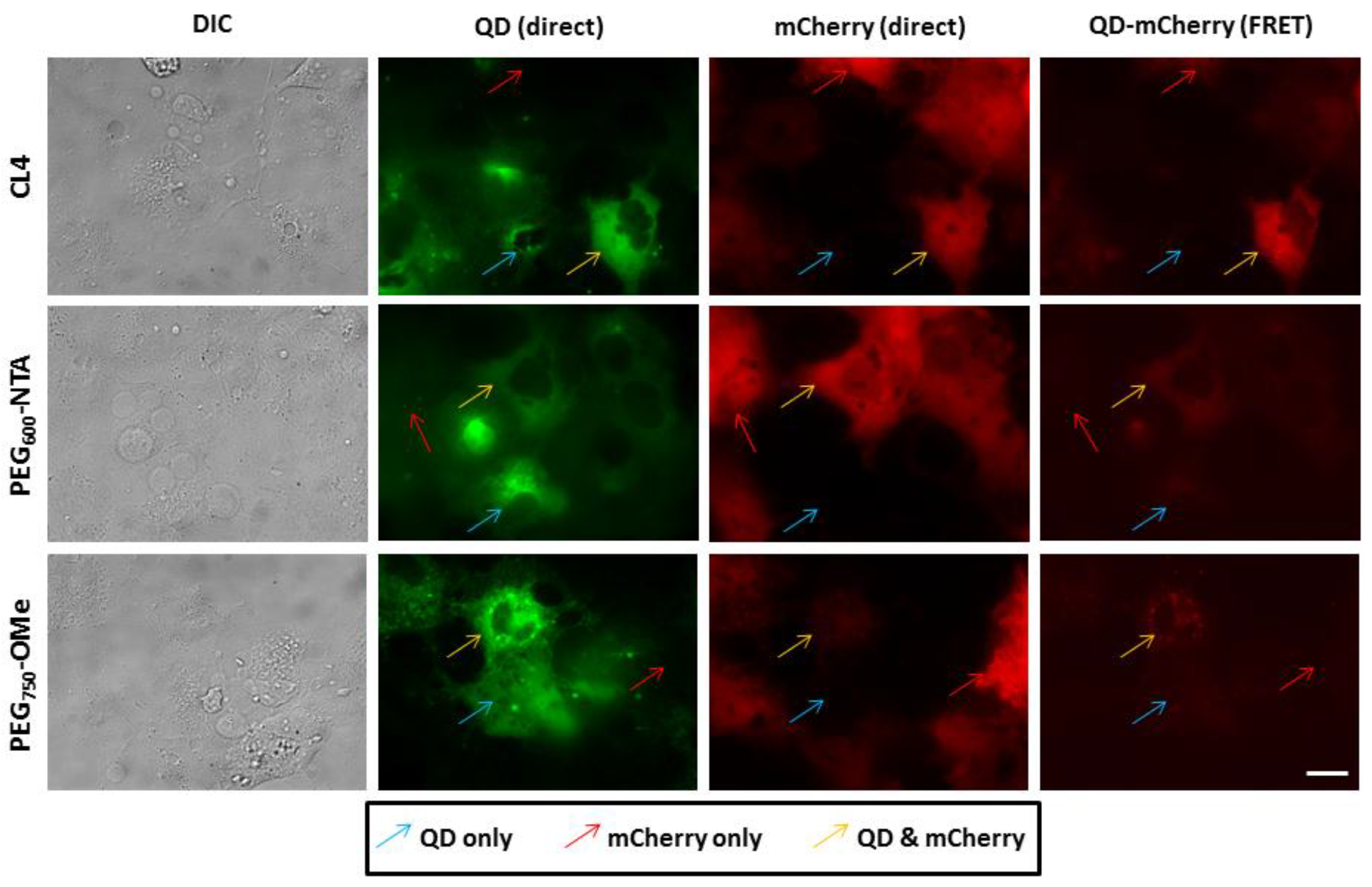
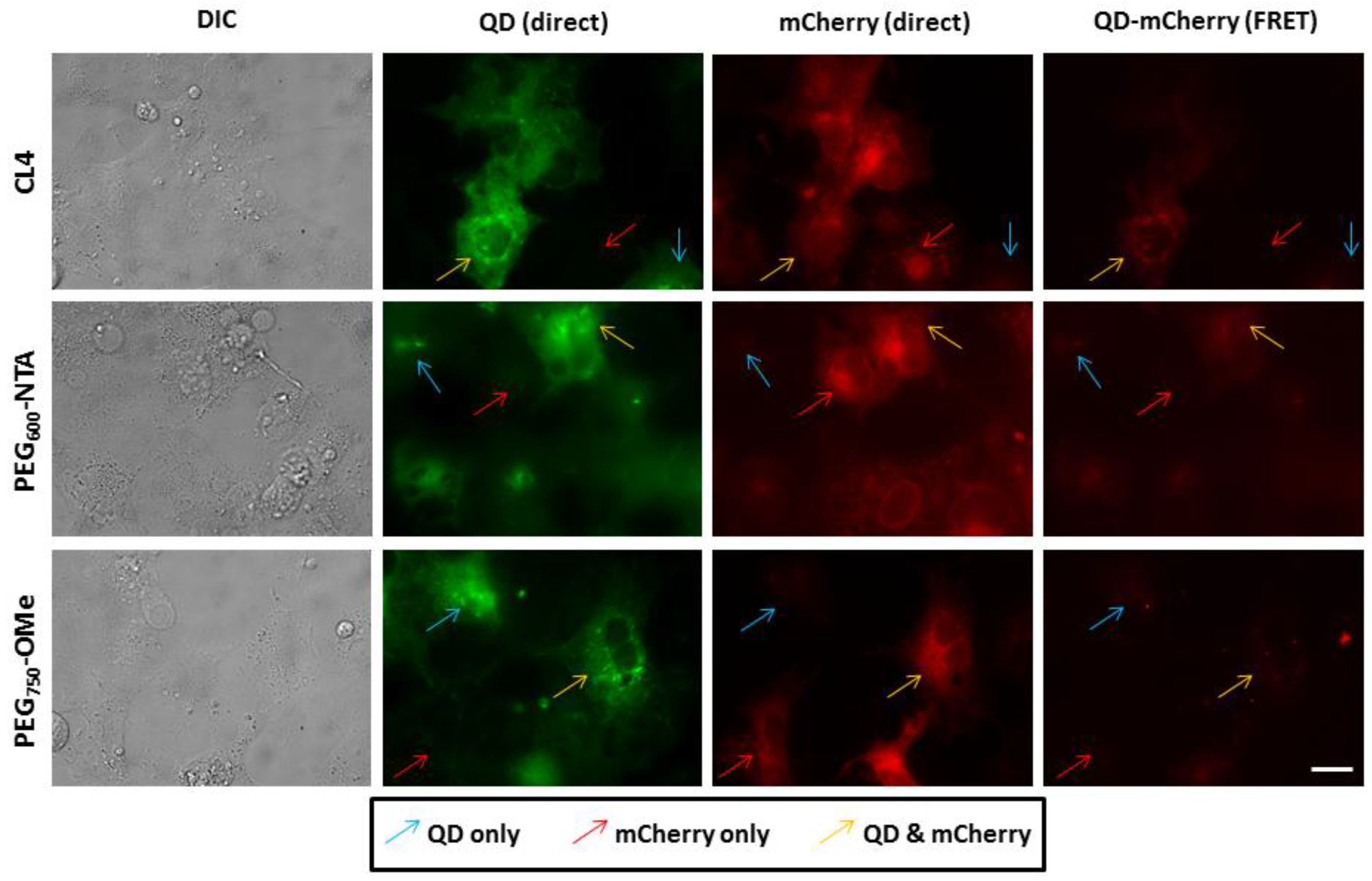
4. Discussion
5. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Dreifuss, T.; Betzer, O.; Shilo, M.; Popovtzer, A.; Motiei, M.; Popovtzer, R. A challenge for theranostics: Is the optimal particle for therapy also optimal for diagnostics? Nanoscale 2015, 7, 15175–15184. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Shin, W.S.; Sunwoo, K.; Kim, W.Y.; Koo, S.; Bhuniya, S.; Kim, J.S. Small conjugate-based theranostic agents: An encouraging approach for cancer therapy. Chem. Soc. Rev. 2015, 44, 6670–6683. [Google Scholar] [CrossRef] [PubMed]
- Goldsmith, C.R.; Jaworski, J.; Sheng, M.; Lippard, S.J. Selective labeling of extracellular proteins containing polyhistidine sequences by a fluorescein-nitrilotriacetic acid conjugate. J. Am. Chem. Soc. 2006, 128, 418–419. [Google Scholar] [CrossRef] [PubMed]
- Guignet, E.G.; Hovius, R.; Vogel, H. Reversible site-selective labeling of membrane proteins in live cells. Nat. Biotechnol. 2004, 22, 440–444. [Google Scholar] [CrossRef] [PubMed]
- Fessenden, J.D. Förster resonance energy transfer measurements of ryanodine receptor type 1 structure using a novel site-specific labeling method. PLoS ONE 2009, 4, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Yip, K.P.; Cha, B.J.; Tse, C.M.; Amin, M.E.; Amin, J. Functional expression of aquaporin-2 tagged with photoconvertible fluorescent protein in mpkccd cells. Cell. Physiol. Biochem. 2015, 36, 670–682. [Google Scholar] [CrossRef] [PubMed]
- Los, G.V.; Encell, L.P.; McDougall, M.G.; Hartzell, D.D.; Karassina, N.; Zimprich, C.; Wood, M.G.; Learish, R.; Ohana, R.F.; Urh, M.; et al. Halotag: A novel protein labeling technology for cell imaging and protein analysis. ACS Chem. Biol. 2008, 3, 373–382. [Google Scholar] [CrossRef] [PubMed]
- Adams, S.R.; Tsien, R.Y. Preparation of the membrane-permeant biarsenicals flash-edt2 and reash-edt2 for fluorescent labeling of tetracysteine-tagged proteins. Nat. Protoc. 2008, 3, 1527–1534. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.J.; Cho, Y.A.; Hwang, H.J.; Kim, J.H. Development of in-cell imaging assay systems for mmp-2 and mmp-9 based on trans-localizing molecular beacon proteins. Arch. Pharm. Res. 2015, 38, 1099–1107. [Google Scholar] [CrossRef] [PubMed]
- Lakowicz, J.R. Principles of Fluorescence Spectroscopy, 3rd ed.; Springer: New York, NY, USA, 2006. [Google Scholar]
- Resch-Genger, U.; Grabolle, M.; Cavaliere-Jaricot, S.; Nitschke, R.; Nann, T. Quantum dots versus organic dyes as fluorescent labels. Nat. Methods 2008, 5, 763–775. [Google Scholar] [CrossRef] [PubMed]
- Dif, A.; Boulmedais, F.; Pinot, M.; Roullier, V.; Baudy-Floćh, M.; Coquelle, F.M.; Clarke, S.; Neveu, P.; Vignaux, F.; le Borgne, R.; et al. Small and stable peptidic pegylated quantum dots to target polyhistidine-tagged proteins with controlled stoichiometry. J. Am. Chem. Soc. 2009, 131, 14738–14746. [Google Scholar] [CrossRef] [PubMed]
- Medintz, I.L.; Pons, T.; Susumu, K.; Boeneman, K.; Dennis, A.; Farrell, D.; Deschamps, J.R.; Melinger, J.S.; Bao, G.; Mattoussi, H. Resonance energy transfer between luminescent quantum dots and diverse fluorescent protein acceptors. J. Phys. Chem. C 2009, 113, 18552–18561. [Google Scholar] [CrossRef] [PubMed]
- Boeneman, K.; Delehanty, J.B.; Susumu, K.; Stewart, M.H.; Medintz, I.L. Intracellular bioconjugation of targeted proteins with semiconductor quantum dots. J. Am. Chem. Soc. 2010, 132, 5975–5977. [Google Scholar] [CrossRef] [PubMed]
- Mercanti, V.; Marchetti, A.; Lelong, E.; Perez, F.; Orci, L.; Cosson, P. Transmembrane domains control exclusion of membrane proteins from clathrin-coated pits. J. Cell Sci. 2010, 123, 3329–3335. [Google Scholar] [CrossRef] [PubMed]
- Mei, B.C.; Susumu, K.; Medintz, I.L.; Delehanty, J.B.; Mountziaris, T.J.; Mattoussi, H. Modular poly(ethylene glycol) ligands for biocompatible semiconductor and gold nanocrystals with extended pH and ionic stability. J. Mater. Chem. 2008, 18, 4949–4958. [Google Scholar] [CrossRef]
- Susumu, K.; Oh, E.; Delehanty, J.B.; Blanco-Canosa, J.B.; Johnson, B.J.; Jain, V.; Hervey, W.J.T.; Algar, W.R.; Boeneman, K.; Dawson, P.E.; et al. Multifunctional compact zwitterionic ligands for preparing robust biocompatible semiconductor quantum dots and gold nanoparticles. J. Am. Chem. Soc. 2011, 133, 9480–9496. [Google Scholar] [CrossRef] [PubMed]
- Susumu, K.; Medintz, I.L.; Delehanty, J.B.; Boeneman, K.; Mattoussi, H. Modification of poly(ethylene glycol)-capped quantum dots with nickel nitrilotriacetic acid and self-assembly with histidine-tagged proteins. J. Phys. Chem. C 2010, 114, 13526–13531. [Google Scholar] [CrossRef]
- Oh, E.; Fatemi, F.K.; Currie, M.; Delehanty, J.B.; Pons, T.; Fragola, A.; Lévêque-Fort, S.; Goswami, R.; Susumu, K.; Huston, A.L.; et al. Pegylated luminescent gold nanoclusters: Synthesis, characterization, bioconjugation, and application to one- and two-photon cellular imaging. Part. Part. Syst. Charact. 2013, 30, 453–466. [Google Scholar] [CrossRef]
- Clapp, A.R.; Medintz, I.L.; Mauro, J.M.; Fisher, B.R.; Bawendi, M.G.; Mattoussi, H. Fluorescence resonance energy transfer between quantum dot donors and dye-labeled protein acceptors. J. Am. Chem. Soc. 2004, 126, 301–310. [Google Scholar] [CrossRef] [PubMed]
- Pons, T.; Uyeda, H.T.; Medintz, I.L.; Mattoussi, H. Hydrodynamic dimensions, electrophoretic mobility, and stability of hydrophilic quantum dots. J. Phys. Chem. B 2006, 110, 20308–20316. [Google Scholar] [CrossRef] [PubMed]
- Prasuhn, D.E.; Deschamps, J.R.; Susumu, K.; Stewart, M.H.; Boeneman, K.; Blanco-Canosa, J.B.; Dawson, P.E.; Medintz, I.L. Polyvalent display and packing of peptides and proteins on semiconductor quantum dots: Predicted versus experimental results. Small 2010, 6, 555–564. [Google Scholar] [CrossRef] [PubMed]
- Delehanty, J.B.; Breger, J.C.; Gemmill, K.B.; Stewart, M.H.; Medintz, I.L. Controlling the actuation of therapeutic nanomaterials: Enabling nanoparticle-mediated drug delivery. Ther. Deliv. 2013, 4, 1411–1429. [Google Scholar] [CrossRef] [PubMed]
- Nazarenus, M.; Zhang, Q.; Soliman, M.G.; del Pino, P.; Pelaz, B.; Carregal-Romero, S.; Rejman, J.; Rothen- Rutishauser, B.; Clift, M.J.; Zellner, R.; et al. In vitro interaction of colloidal nanoparticles with mammalian cells: What have we learned thus far? Beilstein J. Nanotechnol. 2014, 5, 1477–1490. [Google Scholar] [CrossRef] [PubMed]
- Resh, M.D. Palmitoylation of ligands, receptors, and intracellular signaling molecules. Sci. STKE 2006, 2006. [Google Scholar] [CrossRef] [PubMed]
- Sebti, S.M. Protein farnesylation: Implications for normal physiology, malignant transformation, and cancer therapy. Cancer Cell 2005, 7, 297–300. [Google Scholar] [CrossRef] [PubMed]
- Lange, A.; Mills, R.E.; Lange, C.J.; Stewart, M.; Devine, S.E.; Corbett, A.H. Classical nuclear localization signals: Definition, function, and interaction with importin alpha. J. Biol. Chem. 2007, 282, 5101–5105. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Field, L.D.; Walper, S.A.; Susumu, K.; Oh, E.; Medintz, I.L.; Delehanty, J.B. Modulation of Intracellular Quantum Dot to Fluorescent Protein Förster Resonance Energy Transfer via Customized Ligands and Spatial Control of Donor–Acceptor Assembly. Sensors 2015, 15, 30457-30468. https://doi.org/10.3390/s151229810
Field LD, Walper SA, Susumu K, Oh E, Medintz IL, Delehanty JB. Modulation of Intracellular Quantum Dot to Fluorescent Protein Förster Resonance Energy Transfer via Customized Ligands and Spatial Control of Donor–Acceptor Assembly. Sensors. 2015; 15(12):30457-30468. https://doi.org/10.3390/s151229810
Chicago/Turabian StyleField, Lauren D., Scott A. Walper, Kimihiro Susumu, Eunkeu Oh, Igor L. Medintz, and James B. Delehanty. 2015. "Modulation of Intracellular Quantum Dot to Fluorescent Protein Förster Resonance Energy Transfer via Customized Ligands and Spatial Control of Donor–Acceptor Assembly" Sensors 15, no. 12: 30457-30468. https://doi.org/10.3390/s151229810
APA StyleField, L. D., Walper, S. A., Susumu, K., Oh, E., Medintz, I. L., & Delehanty, J. B. (2015). Modulation of Intracellular Quantum Dot to Fluorescent Protein Förster Resonance Energy Transfer via Customized Ligands and Spatial Control of Donor–Acceptor Assembly. Sensors, 15(12), 30457-30468. https://doi.org/10.3390/s151229810