Internal Calibration Förster Resonance Energy Transfer Assay: A Real-Time Approach for Determining Protease Kinetics

Abstract
: Förster resonance energy transfer (FRET) technology has been widely used in biological and biomedical research. This powerful tool can elucidate protein interactions in either a dynamic or steady state. We recently developed a series of FRET-based technologies to determine protein interaction dissociation constant and for use in high-throughput screening assays of SUMOylation. SUMO (small ubiquitin-like modifier) is conjugated to substrates through an enzymatic cascade. This important posttranslational protein modification is critical for multiple biological processes. Sentrin/SUMO-specific proteases (SENPs) act as endopeptidases to process the pre-SUMO or as isopeptidases to deconjugate SUMO from its substrate. Here, we describe a novel quantitative FRET-based protease assay for determining the kinetics of SENP1. Our strategy is based on the quantitative analysis and differentiation of fluorescent emission signals at the FRET acceptor emission wavelengths. Those fluorescent emission signals consist of three components: the FRET signal and the fluorescent emissions of donor (CyPet) and acceptor (YPet). Unlike our previous method in which donor and acceptor direct emissions were excluded by standard curves, the three fluorescent emissions were determined quantitatively during the SENP digestion process from onesample. New mathematical algorithms were developed to determine digested substrate concentrations directly from the FRET signal and donor/acceptor direct emissions. The kinetic parameters, kcat, KM, and catalytic efficiency (kcat/KM) of SENP1 catalytic domain for pre-SUMO1/2/3 were derived. Importantly, the general principles of this new quantitative methodology of FRET-based protease kinetic determinations can be applied to other proteases in a robust and systems biology approach.1. Introduction
SUMO (small ubiquitin-like modifier), a reversible posttranslational protein modifier, is essential in a variety of eukaryotic cellular events, including cell-cycle progression, transcriptional regulation, protein subcellular localizations, protein degradation and chromatin organization [1–5]. Mammalian cells usually express three SUMO paralogs (SUMO1, 2 and 3). A fourth paralog (SUMO4) was discovered in kidney cells and linked to a number of autoimmune diseases, but its biological role is unclear [6,7]. Like ubiquitination, SUMOylation is a dynamic and reversible process. SUMO conjugation occurs through a cascade of reactions that are performed by an activating enzyme (E1), a conjugating enzyme (E2) and, usually, a SUMO ligase (E3) [3,8,9]. The precursor of SUMO peptides needs to be processed by SENP (SUMO-specific protease) before the conjugation cascade [10,11]. The human SENP family of cysteine proteases has six true SENPs (i.e., SENP1-3 and SENP5-7) [10,12,13].
The catalytic efficiency or specificity of an enzyme is best characterized by the ratio of the kinetic constants, kcat/KM. Several biochemical and fluorescent methods are commonly used to examine enzyme kinetics, including the western blotting, radioactive-labeled substrate, a fluorescent-labeled peptide substrate and a fluorescent protein–labeled substrate. All current methods have some limitations and drawbacks. For example, the maturation of pre-SUMO1, 2 and 3 by the catalytic domain of SENP1 was studied in solution and by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) [14,15]. The result showed SENP1's preference but cannot provide a detailed quantitative analysis due to the limitations of slow signal detection of the method. An organic fluorophore, 7-amino-4-carbamoylmethylcoumarin (ACC), was used to label a tetrapeptide substrate (QTGG). The ACC moiety was quenched in the intact substrate but became highly fluorescent upon cleavage by SENPs. Due to the distorted binding activity of small peptide substrate, the kcat/KM determined for SENP1 for the QTGG peptide was ∼300 M−1·s−1, which was up to two orders of magnitude lower than the natural substrates [15,16]. An assay-tagged 7-amino-4-methylcoumarin (AMC) with mature SUMOs was also developed. The values of kcat/KM wwere 2.4 × 106 M−1·s−1 for SUMO1 and 5.6 × 105 M−1·s−1 for SUMO2. These values are generally lower than traditionally considered enzyme kinetics numbers due to the reason noted above. In addition, this system cannot clearly differentiate the isopeptidase and endopeptidase activities of SENPs as there is no specific sequence of either SUMO tail or SUMO-specific substrate after the AMC moiety [17].
Time-resolved FRET assays have been used to characterize the deubiquitinating enzymes (DUBs) or SENPs. The FRET pair Eu-cryptate and allophycocyanin (APC) were tagged to anti-Myc and anti-FLAG antibodies, which interacted with Myc and FLAG on the N- and C-termini of pre-Nedd8, respectively [18]. Terbium (Tb) and YFP (yellow fluorescent protein), another FRET pair, were tagged on SUMO and anti-RanGAP, respectively, to study SUMOylation and SENP's deconjugation [19]. The same FRET pair was tagged onto the N- and C-termini of ubiquitin to study the DUB's processing by the time-resolved FRET (TR-FRET) [20]. However, these assays require additional steps for immune antibodies conjugation or chemical conjugation of thiol-reactive Tb chelate to ubiquitin-AC or other fluorophores. The low conjugation efficiency and denaturation during the conjugation process may lead to inaccurate results of quantitative analysis.
Recent studies focused on efforts to develop quantitative FRET techniques with differentiations of donor, acceptor and net FRET signal to determine molecular interaction events for the protein interaction dissociation constant Kd by calibration curve [21] or direct estimation via direct “cross-talk” ratio measurements [22]. We developed a highly sensitive FRET-based protease assay for determining the kinetic parameters of SENP1 for pre-SUMO1s cleavage with a hybrid substrate, CyPet-preSUMO1-YPet [12]. The absolute FRET signal and direct emissions of the donor and acceptor were determined using two external standard curves (SC) of CyPet-SUMO1 and YPet, and CyPet-SUMO1, YPet and CyPet-preSUMO1-YPet, respectively. The absolute fluorescent signals were converted into protein concentrations by pre-established fluorescent protein standard curves. This method needs several steps to determine absolute FRET signal and convert it to protein concentrations, and during the conversions variability is introduced in each step of measurements.
Here, we report development of a novel quantitative FRET analytic method for determining SENP1 kinetics parameters from one-sample measurement with the “cross-talk” ratio of the donor and acceptor. The fluorescent emissions of the three components (i.e., donor direct emission, acceptor direct emission and absolute FRET emission at acceptor emission wavelength) were monitored during the digestion of a CyPet-PreSUMO-YPet fusion substrate by SENP1. A new mathematical algorithm was developed to directly convert FRET signal to amount of digested substrate. The kinetics parameters, KM, kcat and kcat/KM, were then derived, and their values were compared with those obtained from the standard curve method. The results showed that KM and kcat from the real-time detection method are very close to those obtained from the standard curve method but with many fewer variations. These results suggest that this novel approach could be a very robust and reliable general method for protease kinetics determination.
2. Experimental
2.1. Plasmid Constructs
The open reading frames of the human SUMO and SENP1 genes were amplified by PCR, and the PCR products were cloned into the PCRII-TOPO vector (Life technologies, Carlsbad, CA, USA). After confirming the constructs by sequencing, the cDNAs encoding CyPet-(pre-SUMO1/2/3)-YPet, CyPet-SUMO1/2/3, YPet, and the catalytic domains of SENP1 were cloned into the pET28 (b) vector (Merck KGaA, Germany), engineered with an N-terminal 6xHistidine tag.
2.2. Protein Expression and Purification
Escherichia coli cells of strain BL21 (DE3) were transformed with pET28 vectors encoding CyPet-(pre-SUMO1/2/3)-YPet, CyPet-SUMO1/2/3, YPet, and the catalytic domains of SENP1. The transformed bacteria were grown in 2xYT medium to an optical density of 0.4–0.5 at 600 nm, by induction with 100 μM isopropyl-β-D-thiogalactoside for 16 hours at 25 °C. The 6xhistidine-tagged recombinant proteins were purified from bacterial lysates with nickel agarose affinity chromatography (Qiagen, Valencia, CA, USA) and eluted in 20 mM Tris-HCl, pH 7.4, 50 mM NaCl, 1 mM DTT. Protein purity was examined by SDS-PAGE, and concentrations of the purified proteins were determined by the Bradford assay (ThermoFisher Scientific, Waltham, MA, USA).
2.3. Self-Fluorescence Cross-Talk Ratio Determination
To determine the cross-talk ratio of CyPet and YPet's self-fluorescence, purified CyPet-SUMO1/2/3 and YPet were incubated individually in 37 °C in buffer containing 20 mM Tris-HCl, pH 7.4, 50 mM NaCl, 0.1% (v/v) Tween-20 and 1 mM DTT to a total volume of 80 μL at concentrations of 10, 20, 50, 100, 200, and 500 nM for 10 minutes and added to each well of a 384-well plate (Greiner, glass bottom).
Fluorescent emissions of CyPet at 475 and 530 nm were detected in a fluorescence multi-well plate reader (Flexstation II384, Molecular Devices, Sunnyvale, CA, USA) when excited at 414 nm to determine the cross-talk ratio α; fluorescent emissions of YPet at 530 nm were detected when excited at 414 and 475 nm to determine the cross-talk ratio β. Three samples were repeated for each concentration.
2.4. Establishing Standard Curves
CyPet-(pre-SUMO1/2/3)-YPet was incubated at 37 ° C in low-salt Tris buffer to a total volume of 80 μL and added to each well of a 96-well plate (Greiner, glass bottom). The emission signals at 475 nm were collected after excitation at 414 nm. The concentration of CyPet-(pre-SUMO1/2) was varied from 0.02 to 0.6 μM and of CyPet-(pre-SUMO3) from 0.4 to 3 μM.
CyPet-SUMO1/2/3 and YPet were incubated at 37 °C in a low-salt Tris buffer to a total volume of 80 μl with 1:1 molar ratio and added to each well of a 96-well plate (Greiner, glass bottom). The emission signals at 475 nm were collected after excitation at 414 nm. The concentration of CyPet-SUMO1/2 was varied from 0.02 to 0.2 μM, and of CyPet-(pre-SUMO3) varied from 0.05 to 0.5 μM.
2.5. Protease Kinetics Assay
FRET-based SUMO processing assays were conducted by measuring the emission intensity of CyPet at 475 nm and of YPet at 530 nm when excited at 414 nm in a fluorescence multi-well plate reader (Flexstation II384, Molecular Devices).
CyPet-(pre-SUMO1/2/3)-YPet was incubated with recombinant catalytic domain of SENP1 at 37 °C in buffer containing 20 mM Tris-HCl, pH 7.4, 50 mM NaCl, 0.1% (v/v) Tween-20 and 1 mM DTT to a total volume of 80 μL and added to each well of a 384-well plate (Greiner, glass bottom).
For the kinetics study of CyPet-(pre-SUMO1/2/3)-YPet processed by SENP1, substrate with different concentrations was digested with 0.15 and 4 nM SENP1 individually. Reactions were tested within original 5 min. One phase association model was used to fit the exponential increased reaction velocity. Data were analyzed by the developed method and plotted in GraphPad Prism V software to fit the Michaelis–Menten equation. Five samples were repeated in each concentration.
3. Results
3.1. Design of Internal Calibration Method to Determine Absolute FRET Signal
A FRET fusion substrate with a strong fluorescent signal for SENP1 endopeptidase activity measurement, CyPet-(pre-SUMO1/2/3)-YPet, was constructed. Digestion by SENP1 releases the products CyPet-SUMO1/2/3/ and the C-terminus of SUMO1/2/3-YPet and the FRET signal decreases (Figure 1), corresponding to the amount of digested substrate [23]. In our previous study of SENP1 and pre-SUMO1 digestion, we determined the change in the absolute FRET signal by subtracting the total fluorescence signal from YPet, the direct emission of the digested CyPet-SUMO1, and the emissions of remaining substrate. The emissions of digested YPet and CyPet-SUMO1 and the remaining substrate were determined using two external standard curves of an equal ratio of digested substrates, and digested substrates plus intact substrate, respectively [20]. This approach requires multiple steps to measure the emissions, and variation was introduced from the different assays. Therefore, we sought to determine donor and acceptor direct emissions by taking advantage of our recent development of “cross-talk” ratio methodology for quantitative FRET analysis [22,24].
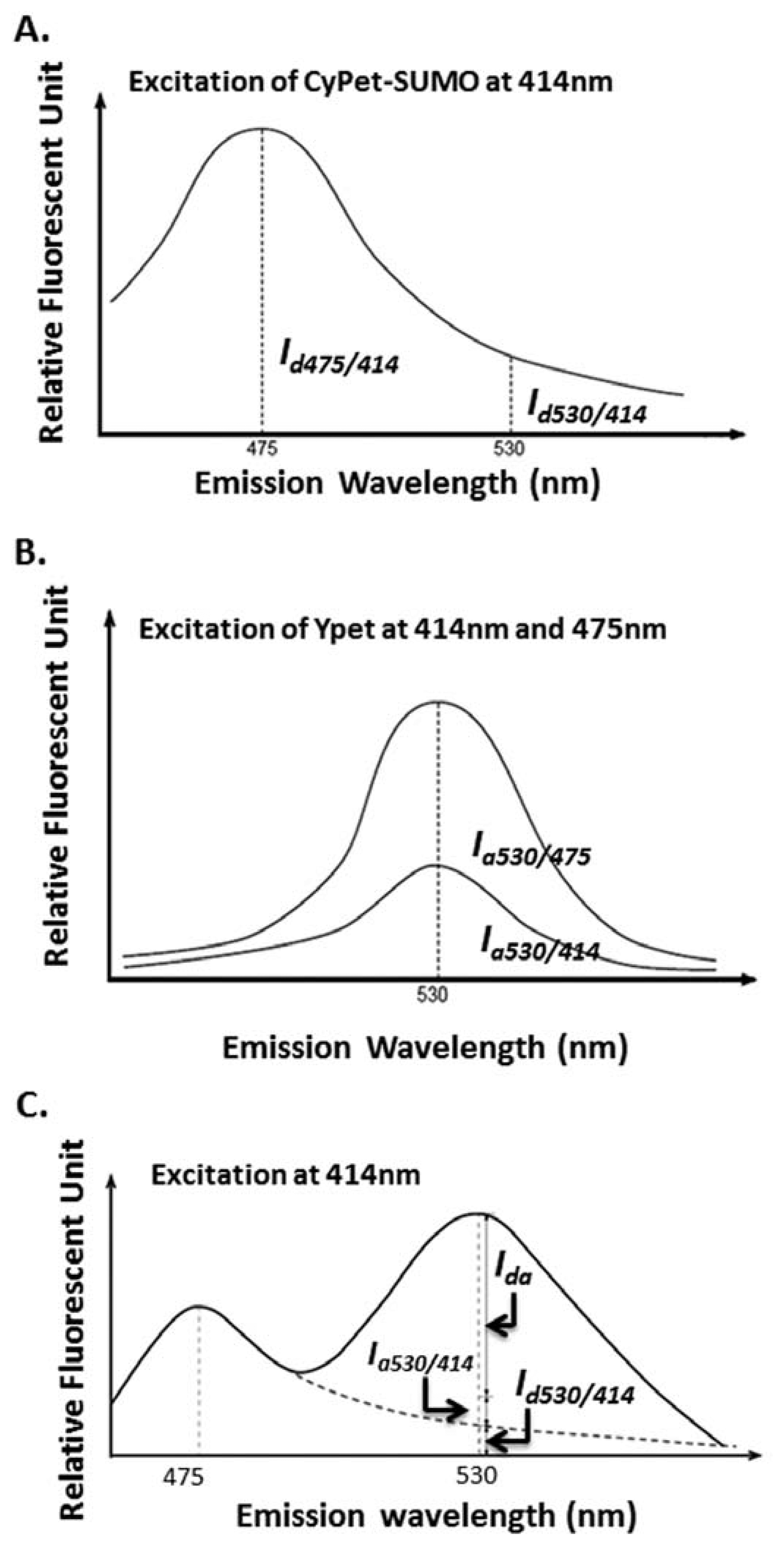
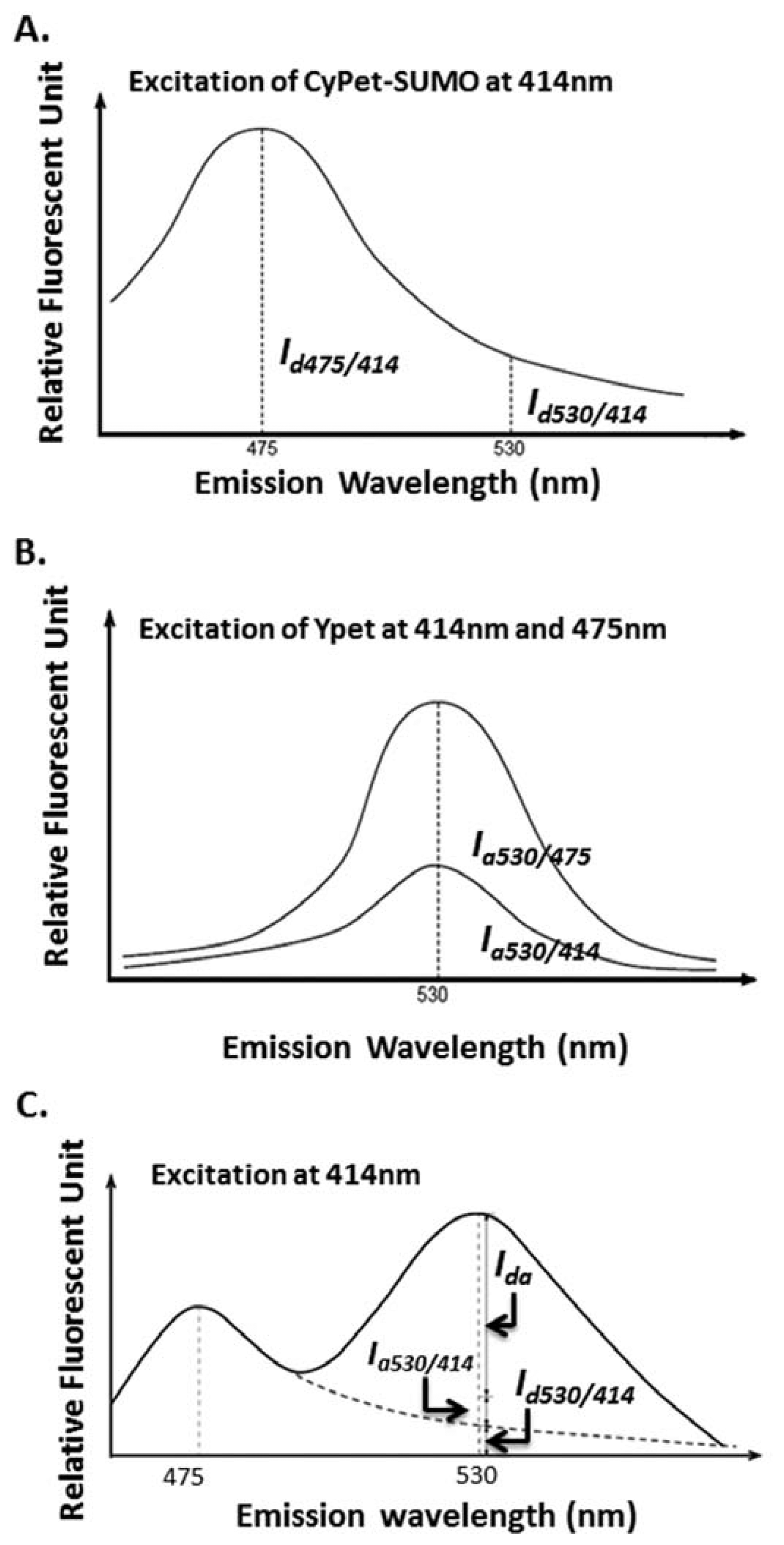
The idea of a “cross-talk” ratio method for a fluorophore direct emission determination is to use two pre-determined ratios for donor and acceptor. The ratios depend on measurements at a different wavelength than the FRET signal emission wavelength (Figure 2(A)) or an emission of acceptor when excited at a wavelength different than FRET excitation wavelength (Figure 2(B)), respectively (Figure 2(C) and see Equations (1–4) below). Then, during the kinetics determination assay, one reaction sample was used to simultaneously determine three emissions, including the total fluorescence emission at the FRET signal emission wavelength, the emission of donor at its maximum emission wavelength of 475 nm when excited at a FRET excitation wavelength of 414 nm, and the emission of acceptor at its maximum emission wavelength of 535 nm when excited at its maximum excitation wavelength of 475 nm. To achieve the highest sensitivity, we chose the maximum emission or excitation wavelengths of donor or acceptor.
3.2. Cross-Talk Ratio Determinations of Cypet-Presumo1/2/3 and Ypet for Absolute FRET Signal Estimation from a One-Sample Measurement
The cross-talk ratio of CyPet's self-fluorescence (α) is the ratio of CyPet-PreSUMO1/2/3's emissions at 530 (Id530/414, and I d as fluorescent signal intensity of donor) to 475 nm (Id475/414) when excited at 414 nm (Figure 2(A)):
We determined the α values of CyPet-PreSUMO1/2/3 for each individual protein. The α values were 0.332, 0.278, and 0.265, respectively. The different values of α for each protein may be due to slight structural differences in the proteins.
The cross-talk ratio of YPet's self-fluorescence (β) is the ratio of YPet's emission at 530 nm when excited at 414 nm (Ia530/414 and Ia as fluorescent signal intensity of acceptor) to the emission at 530 nm when excited at 475 nm (Ia530/475) (Figure 2(B)):
We determined the β value for YPet with purified YPet protein. The value determined for β was 0.026.
Therefore, we differentiated the total fluorescence signal at 530 nm when excited at 414 nm (FL530/414) as the contributions of three components: FRET-induced acceptor's emission (FRET signal or Ida), donor's direct emission(Id530/414) and acceptor's direct emission (Ia530/414) (Figure 2(C)):
According to the definitions of α and β:
After digestion by SENP1, the fluorescent signal at 530 nm decreased, and the fluorescent signal at 475 nm increased because of the disruption of FRET signal after substrate digestion. The remaining fluorescent emission at 530 nm (FL′530/414) could still be divided into a similar three components:
3.3. Determine Digested Substrate Concentration from the FRET Signal Changes, Which Are Determined by Internal Calibration Method
The amount of digested substrate is correlated with the decrease of absolute FRET signal. Therefore, after treatment with SENP1, the remaining FRET-induced acceptor's emission was:
In this newly developed internal calibration (IC) method, I′d475/414 can be directly detected during the experiment. Therefore, the fluorescent signal detected at 530 nm when excited at 414 nm can be expressed as:
During the experiment, and were directly measured by the instrument, Flexstation II386, whereas, FL530/414, Id475/414,Ia530/475, α and β were predetermined before the experiment. So, the two fluorescence signals for determining the amount of the digested substrate are determined during the experiment in real time.
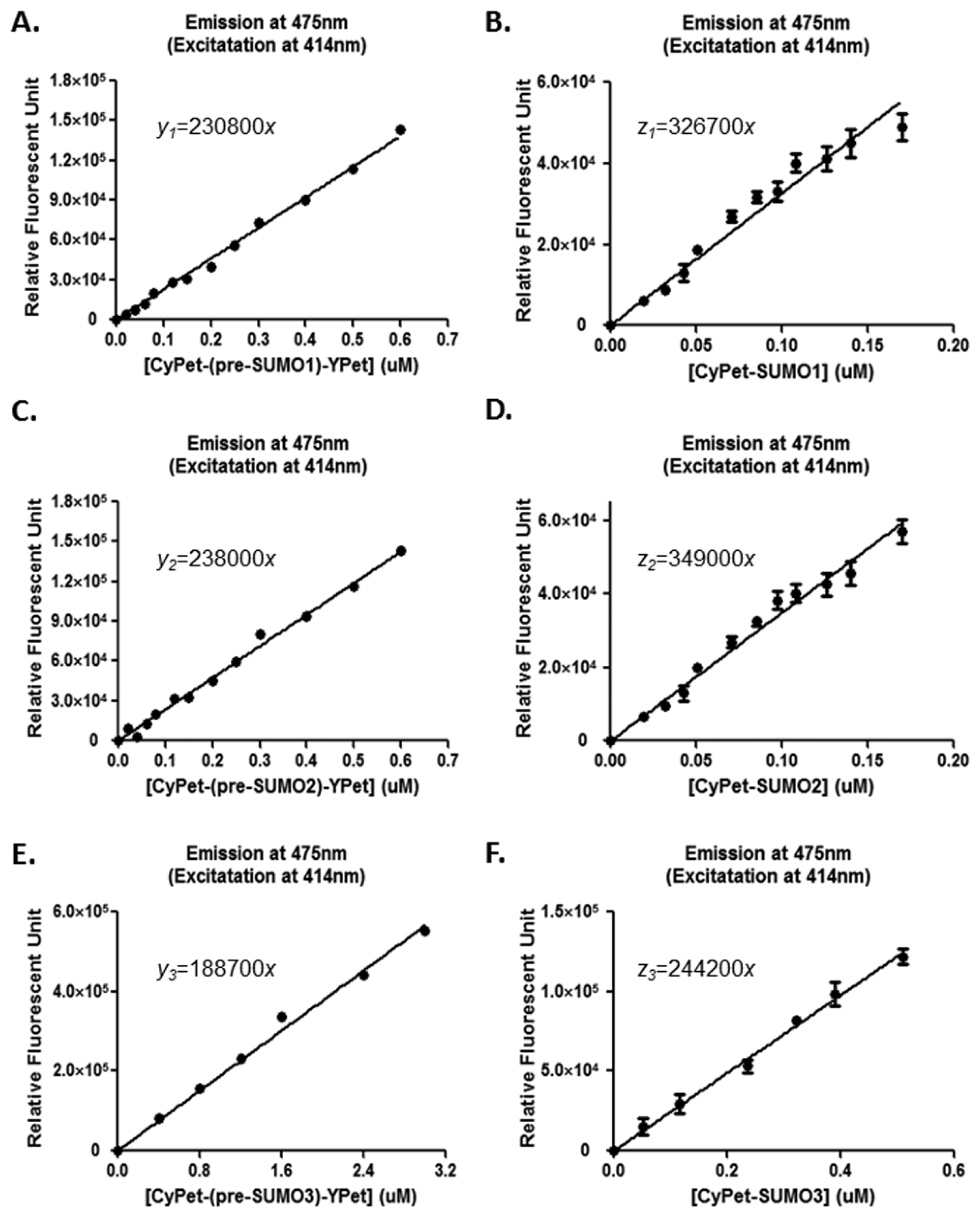
In the previous method [23], standard curves were established to correlate the undigested and digested substrate concentrations to fluorescent signals (Appendix Figure A1). The direct emission of CyPet was determined from both the digested CyPet-SUMO1/2/3 (I′d-cs) and the remaining CyPet-(pre-SUMO1/2/3)-YPet (I′d-csy) substrates:
According to the established standard curves:
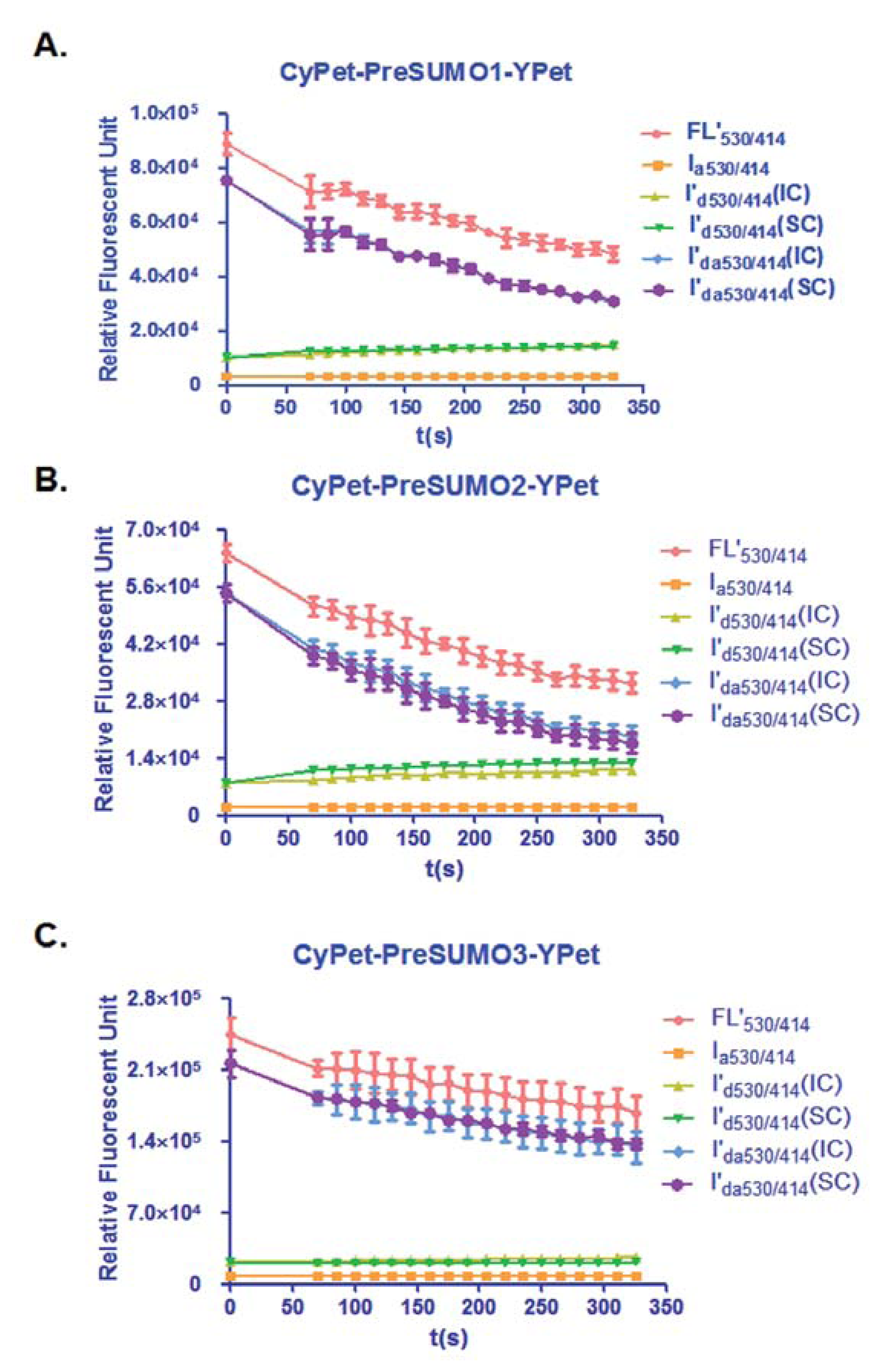
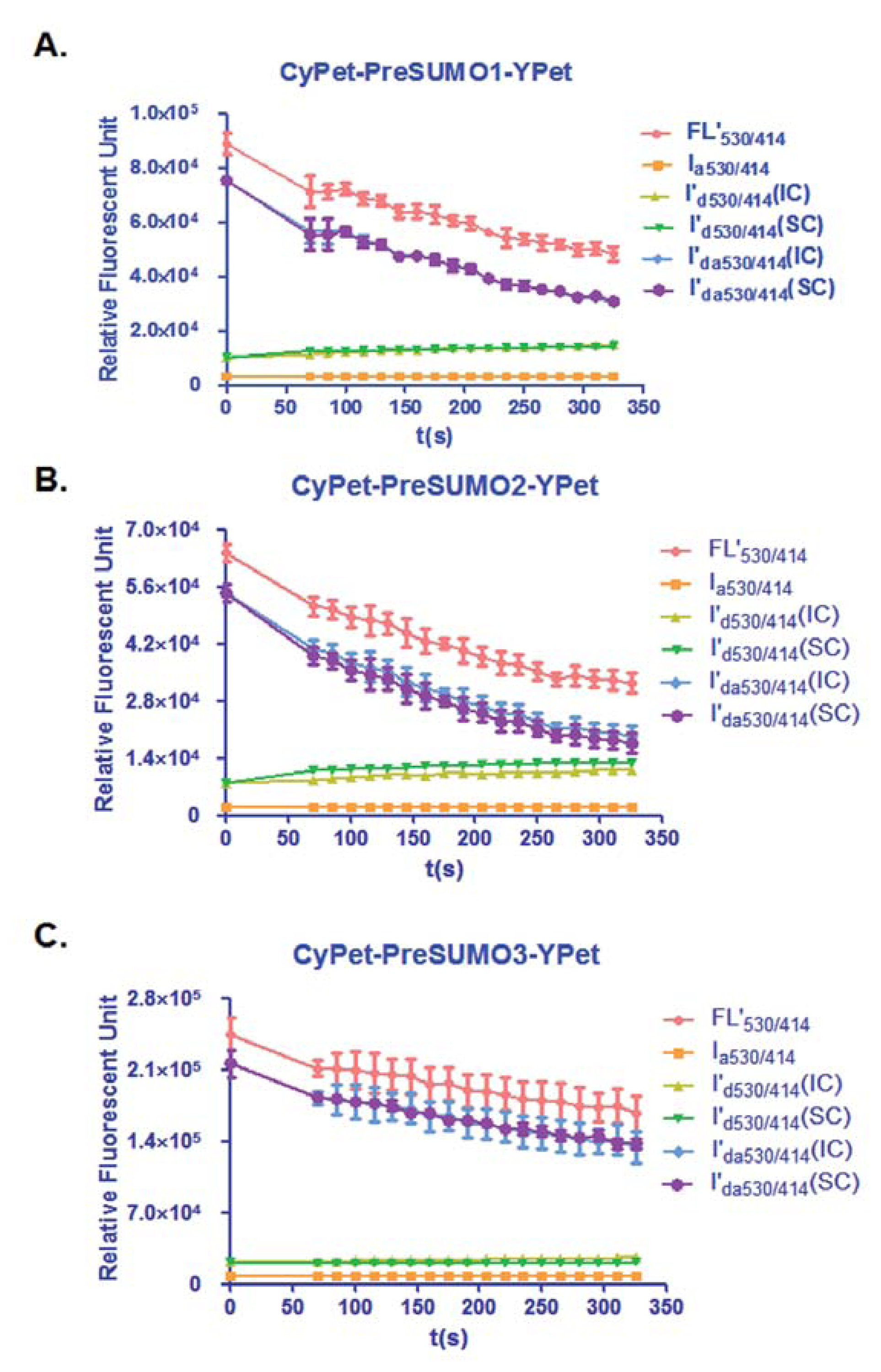
In this study, the total fluorescent emission detected at 530 nm, CyPet and YPet direct emission, as well as the FRET-induced YPet's emission, was analyzed by the internal calibration method and directly compared with those determined by the standard curve method for the concentration of CyPet-(pre-SUMO1)-YPet at 0.15 μM, CyPet-(pre-SUMO2)-YPet at 0.15 μM, and CyPet-(pre-SUMO3)-YPet at 0.4 μM (Figure 3). In all the experiments, the fluorescent signal changes from the internal calibration method were very similar to these determined by the standard curve method, but with significantly less variation and less variation in the final kinetics determinations (see Table 1 below). In addition, the internal calibration method eliminates the need of the standard curves and reduces significant amount experimental procedure.
3.4. Initial Velocity Determination of SENP1 with Pre-SUMO1/2/3 Substrates
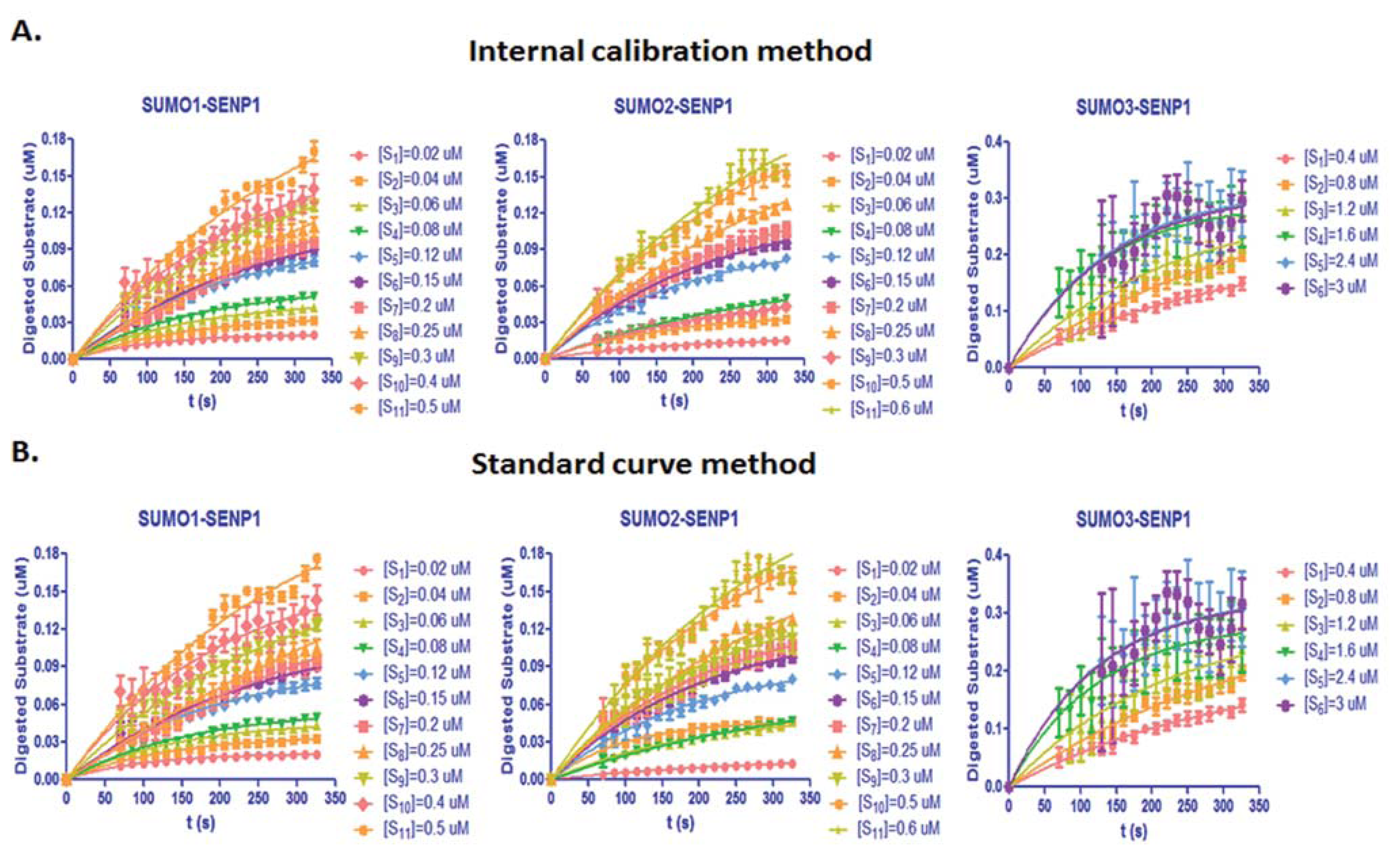
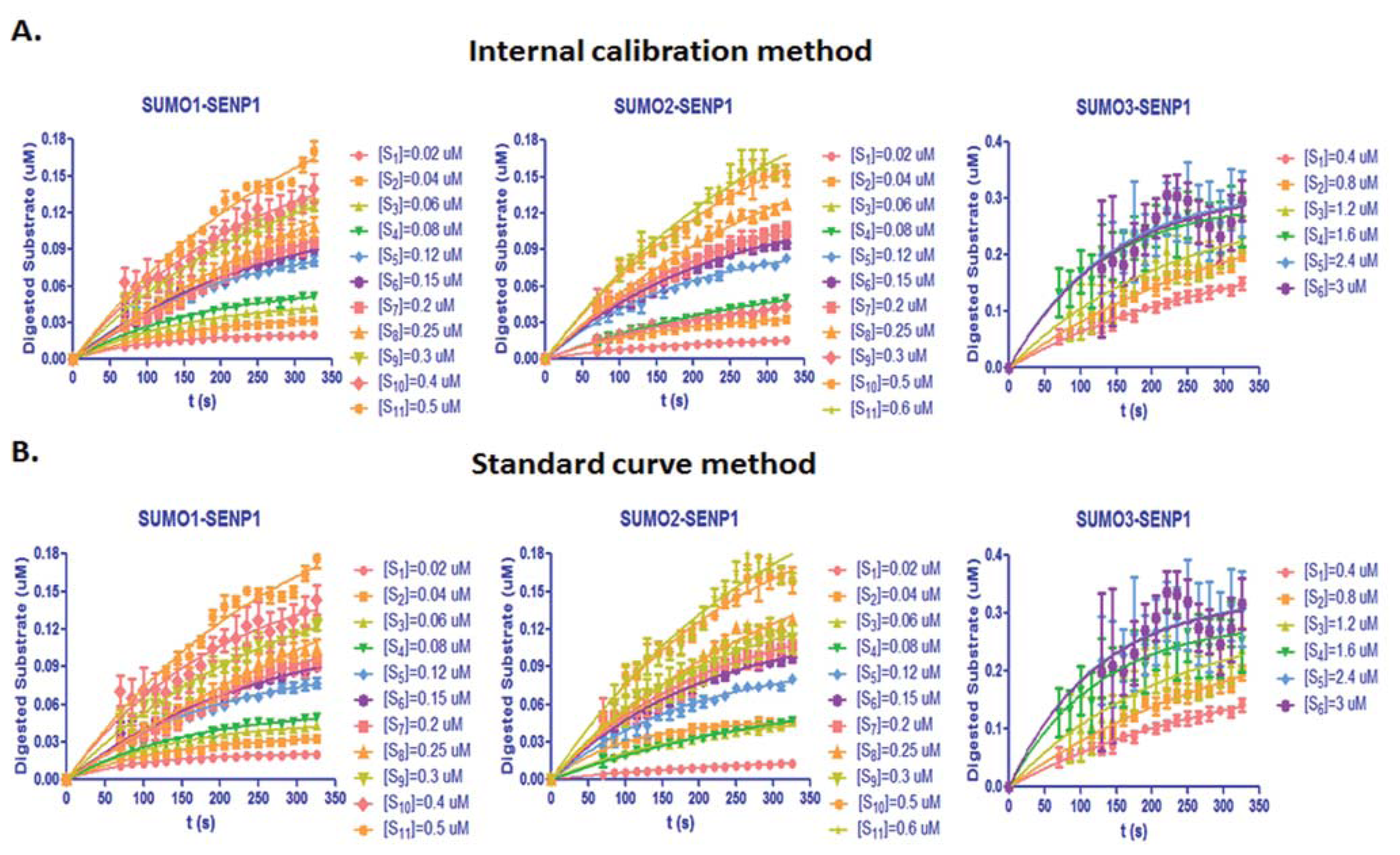
From Equation (7), the digested substrate at different time points can be calculated after the absolute FRET signal changes were determined by the real-time internal calibration method. The amounts of digested substrate, CyPet-(pre-SUMO1/2/3)-YPet, were plotted against the digestion time (Figure 4(A)). The amounts of digested substrates were also determined by the standard curve method (Figure 4(B)). These two methods show similar digestion curves, but with less variation from the internal calibration method. Different amounts of the fluorescent substrates, CyPet-(pre-SUMO1)-YPet (0.02–0.5 μM), CyPet-(pre-SUMO2)-YPet (0.02–0.6 μM), and CyPet-(pre-SUMO3)-YPet (0.4–3.0 μM), were incubated with 0.15 nM (for SENP1/2), and 4 nM (for SENP3) catalytic domain of SENP1, respectively. The emissions at 530 nm and 475 nm, when excited at 414 nm, were determined. A greater substrate concentration for pre-SUMO3 was used because the digestion activity of SENP1 for pre-SUMO3 is much less efficient, and signal changes were very small. This was confirmed later in Kcat/KM determination. The digested substrates, x, were then calculated by the both internal calibration (Figure 4(A)) and the standard curve methods (Figure 4(B)).
The initial velocity (Vo) of CyPet-(pre-SUMO1/2/3)-YPet's maturation by SENP1 was determined as described [23]. Briefly, to determine the reaction velocity of SENP1, the reaction rate (v) was correlated to the change in the amount of substrate (S):
As the digested substrate (or product) concentration increases exponentially from 0 when t = 0, to [S]0 at infinite time:
Accordingly, the original velocity (v0) of CyPet-(pre-SUMO1)-YPet's maturation by SENP1 is:
The results of initial velocity at different concentrations of substrates determined by the two methods are reported in Appendix Table A1. These results also suggest that SENP1 prefers the pre-SUMO1/2 substrates over the pre-SUMO3 substrate. The results displayed a good substrate dose-dependent relationship and showed the small differences from the above two methods.
3.5. Enzyme Kinetics Determination by Michaelis-Menten Analysis
The catalytic specificity and efficiency of an enzyme for a specific substrate are best defined by the ratio of the kinetic constants, kcat/KM. This ratio is generally used to compare the efficiencies of different enzymes towards one substrate or different substrates catalyzed by one enzyme.
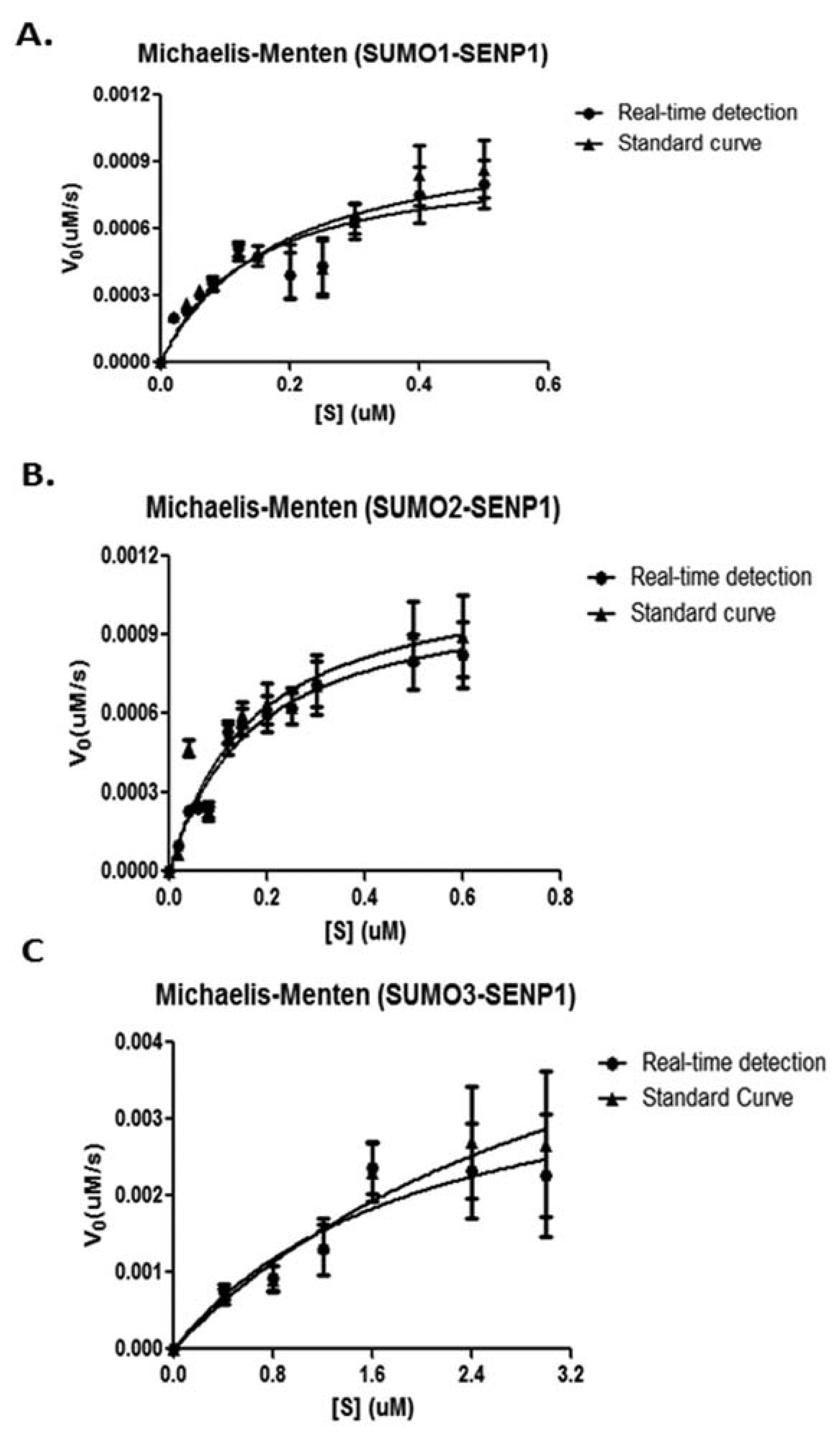
The KM and Vmax values can be obtained from the Michaelis-Menten equation by plotting the initial velocities of SENP1 digestion versus the corresponding concentrations of different substrates. The obtained initial velocities in the Appendix Table A1 were plotted in Michaelis-Menten models for pre-SUMO1/2/3, respectively (Figure 5). The derived values of KM, kcat and kcat/KM ratio were listed in the Table 1.
Compared to a previous study of SENP1's activity by ratiometric method analyzed FRET-based protease assay [25], we obtained similar values for the ratios of kcat/KM of pre-SUMO1 (4.41 × 107vs. 3.92 × 107 M−1·s−1), pre-SUMO2 (4.28 × 107vs. 4.60 × 107 M−1·s−1) and pre-SUMO3 (5.2 × 105vs. 5.95 × 105 M−1·s−1). The values of KM, kcat and kcat/KM ratio for pre-SUMO1 and pre-SUMO2's maturation by SENP1 were very similar. In contrast, the catalytic activity of SENP1 for pre-SUMO3 is much less than the pre-SUMO1 and pre-SUMO2.
4. Discussion
Here, we report a novel quantitative FRET method for determining protease SENP1 kinetics from one-sample measurement in real time for a SUMO systems biology application. In this approach, the absolute FRET signal that is correlated to the digested substrate was continuously determined during the SENP1 digestions of CyPet-pre-SUMO1/2/3-YPet substrate. New mathematic algorithms were developed to differentiate absolute FRET signal from the direct emissions of donor and acceptor at 530 nm, and derive digested substrate from the absolute FRET signal in real time. The direct emission of CyPet at 530 nm, which can be divided into two parts (i.e., undigested CyPet-(pre-SUMOs)-YPet and digested CyPet-SUMOs), was directly obtained from the cross-talk ratio, α. The CyPet emission at 475 nm was determined during digestion in real time, and the values were used to determine the CyPet direct emission at 530 nm. The YPet direct emission at 530 nm was determined by the product of its emission at 530 nm when excited at 475 nm and the cross-talk ratio, β.
In our previous study, the kinetics of pre-SUMO1's maturation by SENP1 was characterized by differentiating and quantifying absolute fluorescent signals contributed by the donor and acceptor emissions and the FRET signal that were then converted into protein concentrations by pre-established fluorescent protein standard curves. The calculated KM, kcat and kcat/KM ratios of these two methods were compared in Table 2. The kcat/KM ratios were similar, but the standard curve method produced greater variation, which came from the errors of standard curves themselves and multiple spectrometer readings. Slopes of standard curves were derived from the linear fitting of fluorescent readings to the protein concentrations. Errors of the slopes cannot be avoided as they result from variations in each protein sample preparation. Nevertheless, the fluorescent emissions for the standard curves were varied slightly from different batches of the recombinant proteins. In addition, the standard curves are needed for each experiment, which is also very tedious. On the other hand, according to a review of different quantitative FRET analytic methods [26], more accurate and robust results can be obtained by observations from multiple channels instead of only one. In this improved approach, real-time fluorescent signals from both 475 and 530 nm were determined, but the previous standard curve method only analyzes the fluorescent signal from 530 nm. Although the derived values for the kinetic parameters were similar, the higher variations form the standard error method indicates that the improved real-time detection method simplifies the measurements by skipping the need of standard curve establishment, and therefore, this method provided more accurate and consistent results.
Pre-SUMO's maturation by SENP1 has been studied by other methods, such as protein gel-based methods and a ratiometric-based FRET assay [14,15,25]. For example, the previous results from gel-based studies showed that SENP1 digests the pre-SUMO1 more efficiently than the pre-SUMO2 and, in turn, more than the pre-SUMO3 [27]. However, the pre-SUMO2 used in the study had an additional nine amino acids after SUMO2's VY tail, and the natural pre-SUMO2 cannot be cleaved in this approach [19]. However, because the gel-based technology is not very quantitative and slow tracking process, the enzymatic kinetics of the maturation process cannot be determined. According to our results, the catalytic activities of SENP1 for pre-SUMO1 and pre-SUMO2 are almost the same in both binding and catalysis steps, and these catalytic activities are much more efficient than for the pre-SUMO3, which is comparable to the previous studies as pre-SUMO1>pre-SUMO3 [20].
Fluorescent proteins can be genetically tagged to interested proteins. CFP (cyan fluorescent protein) and YFP were used as a FRET pair to study the SENP1's endopeptidase activities [25,28]. The kcat/KM determined from the assay was 3.8 × 107 M−1·s−1 for SUMO1 and 5.95 × 105 M−1·s−1 for SUMO3. However, the above FRET-based protease assays used the ratio of acceptor's emission to donor's emission (under the excitation of donor) as the quantitative parameter to characterize the FRET signals without considering the self-fluorescence from donors and acceptors. Although the numerical values of kinetics are similar to ours, this method resulted from an inaccurate FRET signal analysis may lead to wrong conclusions if different FRET pairs are used [26,29].
In summary, we developed a new theoretical and experimental procedure for protease kinetics parameter determination based on quantitative FRET assay of high-sensitive hybrid fluorescent sensor. Except the two pre-determined “cross-talk” ratios of donor and acceptor that should be universal for a specific substrate, all the measurements are carried out from one reaction sample. This approach will significantly shorten experimental procedure and also reduce experimental variations because of minimum experimental steps. This methodology can be applied for any FRET pairs and protease in general.
Acknowledgments
We thank all of the members in Liao's group for very close collaborative work and for help with this work. This work was supported by the National Institutes of Health (Grant AI076504 to Jiayu Liao).
Appendix


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Table A1-1. Initial velocity determinations of SENP1 for SUMO1. | ||
|---|---|---|
| [S[(μM) | Vo(μM/s) | |
| Real-Time Detection | Standard Curve | |
| 0.02 | 0.00020 ± 0.000010 | 0.00021 ± 0.000010 |
| 0.04 | 0.00023 ± 0.000014 | 0.00026 ± 0.000016 |
| 0.06 | 0.00030 ± 0.000017 | 0.00033 ± 0.000018 |
| 0.08 | 0.00035 ± 0.000029 | 0.00035 ± 0.000028 |
| 0.12 | 0.00051 ± 0.000033 | 0.00050 ± 0.000037 |
| 0.15 | 0.00048 ± 0.000044 | 0.00048 ± 0.000044 |
| 0.2 | 0.00039 ± 0.000010 | 0.00041 ± 0.000012 |
| 0.25 | 0.00044 ± 0.000013 | 0.00042 ± 0.000013 |
| 0.30 | 0.00064 ± 0.000069 | 0.00063 ± 0.000079 |
| 0.50 | 0.00075 ± 0.000013 | 0.00084 ± 0.000013 |
| 0.60 | 0.00080 ± 0.000011 | 0.00087 ± 0.000013 |
| Table A1-2. Initial velocity determinations of SENP1 for SUMO1 | ||
|---|---|---|
| [S[(μM) | Vo(μM/s) | |
| Real-time detection | Standard curve | |
| 0.02 | 0.000096 ± 0.000007 | 0.000065 ± 0.000008 |
| 0.04 | 0.00023 ± 0.000022 | 0.00047 ± 0.000031 |
| 0.06 | 0.00024 ± 0.000020 | 0.00026 ± 0.000023 |
| 0.08 | 0.00024 ± 0.000031 | 0.00022 ± 0.000028 |
| 0.12 | 0.00053 ± 0.000043 | 0.00050 ± 0.000056 |
| 0.15 | 0.00057 ± 0.000049 | 0.00060 ± 0.000048 |
| 0.2 | 0.00060 ± 0.000068 | 0.00064 ± 0.000081 |
| 0.25 | 0.00062 ± 0.000059 | 0.00063 ± 0.000068 |
| 0.3 | 0.00071 ± 0.000088 | 0.00071 ± 0.000011 |
| 0.5 | 0.00080 ± 0.000011 | 0.00091 ± 0.000012 |
| 0.6 | 0.00082 ± 0.000013 | 0.00090 ± 0.000016 |
| Table A1-3. Initial velocity determinations of SENP1 for SUMO1. | ||
|---|---|---|
| [S[(μM) | Vo(μM/s) | |
| Real-time detection | Standard curve | |
| 0.4 | 0.00075 ± 0.000094 | 0.00068 ± 0.000010 |
| 0.8 | 0.00093 ± 0.000016 | 0.00091 ± 0.000017 |
| 1.2 | 0.0013 ± 0.00033 | 0.00013 ± 0.000037 |
| 1.6 | 0.0024 ± 0.00034 | 0.00027 ± 0.000073 |
| 2.4 | 0.0023 ± 0.00062 | 0.00027 ± 0.000095 |
| 3.0 | 0.0023 ± 0.00080 | 0.00027 ± 0.00016 |
Conflict of Interest
The authors declare that they have no competing financial interests.
References
- Hannoun, Z.; Greenhough, S.; Jaffray, E.; Hay, R.T.; Hay, D.C. Post-Translational modification by SUMO. Toxicology 2010, 278, 288–293. [Google Scholar]
- Lomelí, H.; Vázquez, M. Emerging roles of the SUMO pathway in development. Cell. Mol. Life Sci. 2011, 68, 4045–4064. [Google Scholar]
- Johnson, E.S. Proteinmodification Bysumo. Annu. Rev. Biochem. 2004, 73, 355–382. [Google Scholar]
- Geiss-Friedlander, R.; Melchior, F. Concepts in sumoylation: A decade on. Nat. Rev. Mol. Cell Biol. 2007, 8, 947–956. [Google Scholar]
- Gill, G. SUMO and ubiquitin in the nucleus: Different functions, similar mechanisms? Genes Develop. 2004, 18, 2046–2059. [Google Scholar]
- Guo, D.; Li, M.; Zhang, Y.; Yang, P.; Eckenrode, S.; Hopkins, D.; Zheng, W.; Purohit, S.; Podolsky, R.H.; Muir, A.; et al. A functional variant of SUMO4, a new I kappa B alpha modifier, is associated with type 1 diabetes. Nat. Genet. 2004, 36, 837–841. [Google Scholar]
- Wang, C.Y.; She, J.X. SUMO4 and its role in type 1 diabetes pathogenesis. Diabetes Metab. Res. Rev. 2008, 24, 93–102. [Google Scholar]
- Kerscher, O.; Felberbaum, R.; Hochstrasser, M. Modification of proteins by ubiquitin and ubiquitin-like proteins. Annu. Rev. Cell Dev. Biol. 2006, 22, 159–180. [Google Scholar]
- Capili, A.D.; Lima, C.D. Taking it step by step: mechanistic insights from structural studies of ubiquitin/ubiquitin-like protein modification pathways. Curr. Opin. Struct. Biol. 2007, 17, 726–735. [Google Scholar]
- Yeh, E.T. SUMOylation and De-SUMOylation: Wrestling with life's processes. J. Biol. Chem. 2009, 284, 8223–8227. [Google Scholar]
- Reyes-Turcu, F.E.; Ventii, K.H.; Wilkinson, K.D. Regulation and cellular roles of ubiquitin-specific deubiquitinating enzymes. Annu. Rev. Biochem. 2009, 78, 363–397. [Google Scholar]
- Drag, M.; Salvesen, G.S. DeSUMOylating enzymes-SENPs. IUBMB Life 2008, 60, 734–742. [Google Scholar]
- Mukhopadhyay, D.; Dasso, M. Modification in reverse: The SUMO proteases. Trends Biochem. Sci. 2007, 32, 286–295. [Google Scholar]
- Xu, Z.; Au, S.W.N. Mapping residues of SUMO precursors essential in differential maturation by SUMO-specific protease, SENP1. Biochem. J. 2005, 386, 325–330. [Google Scholar]
- Mikolajczyk, J.; Drag, M.; Békés, M.; Cao, J.T.; Ronai, Z.; Salvesen, G.S. Small ubiquitin-related modifier (SUMO)-specific proteases: Profiling the specificities and activities of human SENPs. J. Biol. Chem. 2007, 282, 26217–26224. [Google Scholar]
- Drag, M.; Mikolajczyk, J.; Krishnakumar, I.M.; Huang, Z.; Salvesen, G.S. Activity profiling of human deSUMOylating enzymes (SENPs) with synthetic substrates suggests an unexpected specificity of two newly characterized members of the family. Biochem. J. 2008, 409, 461–469. [Google Scholar]
- Kolli, N.; Mikolajczyk, J.; Drag, M.; Mukhopadhyay, D.; Moffatt, N.; Dasso, M.; Salvesen, G.; Wilkinson, K.D. Distribution and paralogue specificity of mammalian deSUMOylating enzymes. Biochem. J. 2010, 430, 335–344. [Google Scholar]
- Engels, I.; Daguia, C.; Huynh, T.; Urbina, H.; Buddenkotte, J.; Schumacher, A.; Caldwell, J.; Brinker, A. A time-resolved fluorescence resonance energy transfer-based assay for DEN1 peptidase activity. Anal. Biochem. 2009, 390, 85–87. [Google Scholar]
- Carlson, C.B.; Horton, R.A.; Vogel, K.W. A Toolbox Approach to High-Throughput TR-FRET-Based SUMOylation and DeSUMOylation Assays. ASSAY and Drug Dev. Technol. 2009, 7, 348–355. [Google Scholar]
- Horton, R.A.; Strachan, E.A.; Vogel, K.W.; Riddle, S.M. A substrate for deubiquitinating enzymes based on time-resolved fluorescence resonance energy transfer between terbium and yellow fluorescent protein. Anal. Biochem. 2007, 360, 138–143. [Google Scholar]
- Martin, S.; Tatham, M.H.; Hay, R.T.; Samuel, D.W. Quantitative analysis of multi-protein interactions using FRET: Application to the SUMO pathway. Protein Sci. 2008, 17, 777–784. [Google Scholar]
- Song, Y.; Madahar, V.; Liao, J. Development of FRET Assay into quantitative and high-throughput screening Technology platforms for protein–protein interactions. Ann. Biomed. Eng. 2011, 39, 1224–1234. [Google Scholar]
- Liu, Y.; Song, Y.; Madahar, V.; Liao, J. Quantitative Förster resonance energy transfer analysis for kinetic determinations of SUMO-specific protease. Anal. Biochem. 2012, 422, 14–21. [Google Scholar]
- Song, Y.; Rodgers, V.G.; Schultz, J.S.; Liao, J. Protein interaction affinity determination by quantitative FRET technology. Biotechnol. Bioeng. 2012, 109, 2875–2883. [Google Scholar]
- Shen, L.; Tatham, M.H.; Dong, C.; Zagórska, A.; Naismith, J.H.; Hay, R.T. SUMO protease SENP1 induces isomerization of the scissile peptide bond. Nat. Struct. Mol. Biol. 2006, 13, 1069–1077. [Google Scholar]
- Berney, C.; Danuser, G. FRET or no FRET: A quantitative comparison. Biophys. J. 2003, 84, 3992–4010. [Google Scholar]
- Shen, L.N.; Dong, C.; Liu, H.; Naismith, J.H.; Hay, R.T. The structure of SENP1–SUMO-2 complex suggests a structural basis for discrimination between SUMO paralogues during processing. Biochem. J. 2006, 397, 279–288. [Google Scholar]
- Martin, S.F.; Hattersley, N.; Samuel, I.D.; Hay, R.T.; Tatham, M.H. A fluorescence-resonance-energy-transfer-based protease activity assay and its use to monitor paralog-specific small ubiquitin-like modifier processing. Anal. Biochem. 2007, 363, 83–90. [Google Scholar]
- Gordon, G.W.; Berry, G.; Liang, X.H.; Levine, B.; Herman, B. Quantitative fluorescence resonance energy transfer measurements using fluorescence microscopy. Biophys. J. 1998, 74, 2702–2713. [Google Scholar]
| Abbreviations | |
|---|---|
| Ubl | Ubiquitin-like protein |
| SUMO | Small ubiquitin-like modifier |
| SENP | SUMO/Sentrin specific protease |
| SDS-PAGE | Sodium dodecyl sulfate-polyacrylamide gel electrophoresis |
| ACC | 7-Amino-4-carbamoylmetylcoumarin |
| AMC | 7-Amino-4-methylcoumarin |
| FRET | Förester resonance energy transfer |
| DUB | Deubiquitinating enzyme |
| CFP | Cyan fluorescent protein |
| YFP | Yellow fluorescent protein |
| DTT | Dithiothreitol |

| FRET signal Analysis method | Km(μM) | Kcat(S−1) | Kcat/Km(M−1·S−1) | ||
|---|---|---|---|---|---|
| SUMO1 | Real-time detection | 0.14 ± 0.05 | 6.14 ± 0.89 | (4.41 ± 1.74) × 107 | |
| Standard curve | 0.18 ± 0.08 | 7.07 ± 1.33 | (3.92 ± 1.85) × 107 | ||
| SUMO2 | Real-time detection | 0.17 ± 0.003 | 7.17 ± 0.54 | (4.28 ± 0.83) × 107 | |
| Standard curve | 0.17 ± 0.01 | 7.66 ± 1.04 | (4.60 ± 1.66) × 107 | ||
| SUMO3 | Real-time detection | 1.99 ± 1.25 | 1.03 ± 0.33 | (5.17 ± 3.65) × 105 | |
| Standard curve | 3.75 ± 2.22 | 1.61 ± 0.62 | (4.31 ± 3.04) × 105 | ||
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Jiang, L.; Liu, Y.; Song, Y.; Saavedra, A.N.; Pan, S.; Xiang, W.; Liao, J. Internal Calibration Förster Resonance Energy Transfer Assay: A Real-Time Approach for Determining Protease Kinetics. Sensors 2013, 13, 4553-4570. https://doi.org/10.3390/s130404553
Jiang L, Liu Y, Song Y, Saavedra AN, Pan S, Xiang W, Liao J. Internal Calibration Förster Resonance Energy Transfer Assay: A Real-Time Approach for Determining Protease Kinetics. Sensors. 2013; 13(4):4553-4570. https://doi.org/10.3390/s130404553
Chicago/Turabian StyleJiang, Ling, Yan Liu, Yang Song, Amanda N. Saavedra, Songqin Pan, Wensheng Xiang, and Jiayu Liao. 2013. "Internal Calibration Förster Resonance Energy Transfer Assay: A Real-Time Approach for Determining Protease Kinetics" Sensors 13, no. 4: 4553-4570. https://doi.org/10.3390/s130404553
APA StyleJiang, L., Liu, Y., Song, Y., Saavedra, A. N., Pan, S., Xiang, W., & Liao, J. (2013). Internal Calibration Förster Resonance Energy Transfer Assay: A Real-Time Approach for Determining Protease Kinetics. Sensors, 13(4), 4553-4570. https://doi.org/10.3390/s130404553