NGS-Based Genotyping, High-Throughput Phenotyping and Genome-Wide Association Studies Laid the Foundations for Next-Generation Breeding in Horticultural Crops



Abstract
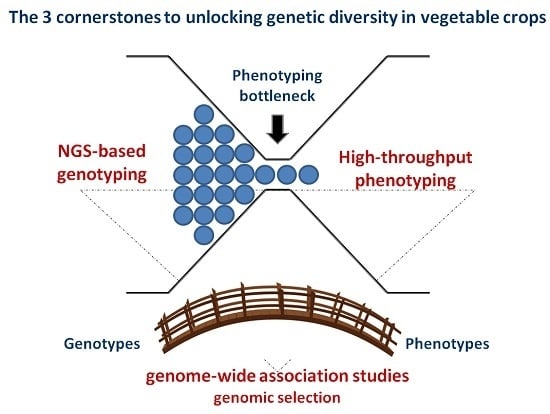
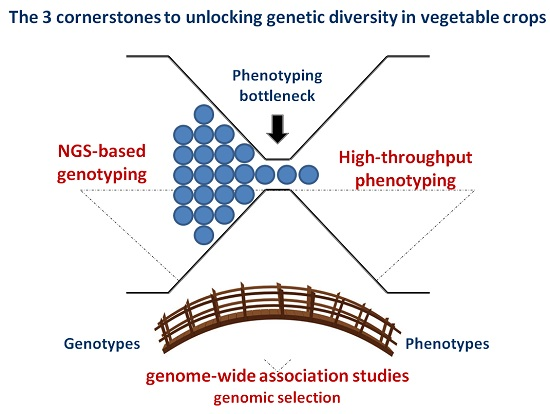{kind=link}
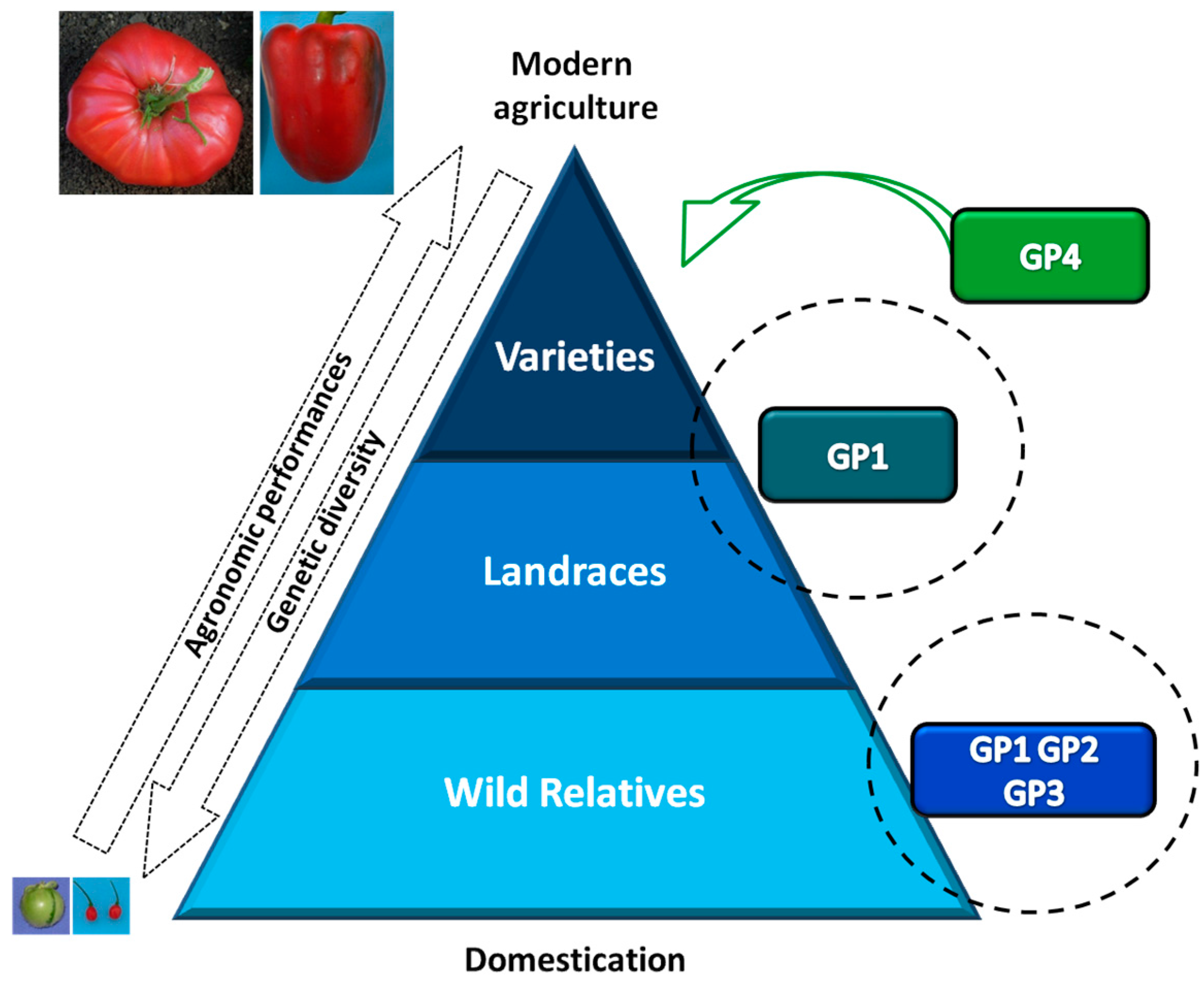{kind=link}
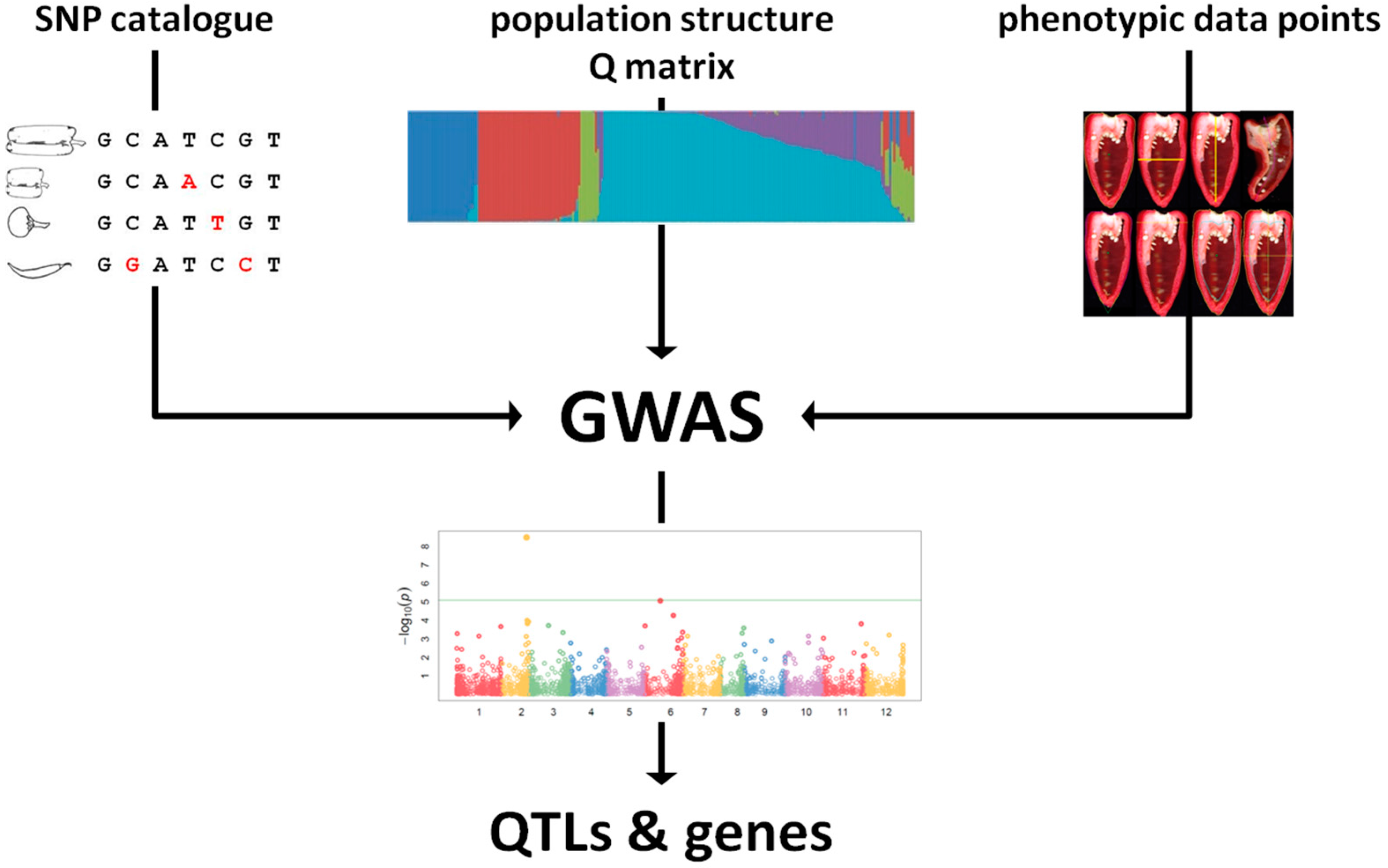{kind=link}
{kind=link}
1. The Use of Plant Genetic Resources in Vegetable Crop Improvement
1.1. Erosion of Genetic Diversity in Crops
1.2. Strategies for Collection and Conservation of Plant Genetic Resources
1.3. Importance of Plant Genetic Resources and Biodiversity in Breeding Programs
2. NGS-Based Genotyping for Genetic Diversity Evaluation
3. Advanced Phenomics in Plant Breeding
4. Linking Genotype to Phenotype
5. Outlook
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| AmpSeq | amplicon sequencing |
| CBD | convention on biological diversity |
| CC | core collections |
| CIR | color infrared |
| CWR | crop wild relatives |
| EMS | ethylmethane sulfonate |
| eQTL | expression QTL |
| FAO | food and agriculture organization of the United Nations |
| FBP | field-based phenotyping |
| FDR | false discovery rate |
| FIGS | focused identification of germplasm strategy |
| GBS | genotype-by-sequencing |
| GEBV | genomic estimated breeding values |
| GIS | geographic information system |
| GLM | general linear model |
| GP | gene pools |
| GS | genomic selection |
| GWAS | genome-wide association studies |
| HS | hyperspectral |
| HTPP | high-throughput plant phenotyping |
| IBLs | inbred backcross lines |
| ILs | introgression lines |
| LAI | leaf area index |
| LD | linkage disequilibrium |
| LR | landraces |
| MAGIC | multi-parent advanced generation inter-cross |
| meQTL | methylation QTL |
| MLM | mixed linear model |
| MRI | magnetic resonance imagers |
| MS | multispectral |
| NDVI | normalized difference vegetation index |
| NGS | next generation sequencing |
| NPBT | novel plant breeding techniques |
| PCR | polymerase chain reaction |
| PGR | plant genetic resources |
| QTN | quantitative trait nucleotides |
| QTL | quantitative trait loci |
| RAD-Seq | restriction site-associated DNA sequencing |
| REs | restriction enzymes |
| RILs | recombinant inbred lines |
| SMD | sterility mosaic disease |
| SNPs | single nucleotide polymorphisms |
| SSR | simple sequence repeat |
| TP | training population |
References
- Meyer, R.S.; Purugganan, M.D. Evolution of crop species: Genetics of domestication and diversification. Nat. Rev. Genet. 2013, 14, 840–852. [Google Scholar] [CrossRef] [PubMed]
- Gepts, P. Crop domestication as a long-term selection experiment. In Plant Breeding Reviews; John Wiley & Sons, Inc.: Oxford, UK, 2010; pp. 1–44. [Google Scholar]
- Meyer, R.S.; DuVal, A.E.; Jensen, H.R. Patterns and processes in crop domestication: An historical review and quantitative analysis of 203 global food crops. New Phytol. 2012, 196, 29–48. [Google Scholar] [CrossRef] [PubMed]
- Flint-Garcia, S.A. Genetics and consequences of crop domestication. J. Agric. Food Chem. 2013, 61, 8267–8276. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.; Zhu, G.; Zhang, J.; Xu, X.; Yu, Q.; Zheng, Z.; Zhang, Z.; Lun, Y.; Li, S.; Wang, X.; et al. Genomic analyses provide insights into the history of tomato breeding. Nat. Genet. 2014, 46, 1220–1226. [Google Scholar] [CrossRef] [PubMed]
- Tieman, D.; Zhu, G.; Resende, M.F.; Lin, T.; Nguyen, C.; Bies, D.; Rambla, J.L.; Beltran, K.S.O.; Taylor, M.; Zhang, B.; et al. A chemical genetic roadmap to improved tomato flavor. Science 2017, 355, 391–394. [Google Scholar] [CrossRef] [PubMed]
- Massawe, F.; Mayes, S.; Cheng, A. Crop diversity: An unexploited treasure trove for food security. Trends Plant. Sci. 2016, 21, 365–368. [Google Scholar] [CrossRef] [PubMed]
- Diamond, J. Evolution, consequences and future of plant and animal domestication. Nature 2002, 418, 700–707. [Google Scholar] [CrossRef] [PubMed]
- Gornall, R.J. Recombination systems and plant domestication. Biol. J. Linn. Soc. 1983, 20, 375–383. [Google Scholar] [CrossRef]
- Brozynska, M.; Furtado, A.; Henry, R.J. Genomics of crop wild relatives: Expanding the gene pool for crop improvement. Plant Biotechnol. J. 2016, 14, 1070–1085. [Google Scholar] [CrossRef] [PubMed]
- Roberts, M.J.; Schlenker, W. World supply and demand of food commodity calories. Am. J. Agric. Econ. 2009, 91, 1235–1242. [Google Scholar] [CrossRef]
- World Population Ageing. Available online: http://www.un.org/en/development/desa/population/publications/pdf/ageing/WorldPopulationAgeing2013 (accessed on 24 July 2017).
- Godfray, H.C.J.; Beddington, J.R.; Crute, I.R.; Haddad, L.; Lawrence, D.; Muir, J.F.; Pretty, J.; Robinson, S.; Thomas, S.M.; Toulmin, C. Food security: The challenge of feeding 9 billion people. Science 2010, 327, 812–818. [Google Scholar] [CrossRef] [PubMed]
- Khoury, C.K.; Bjorkman, A.D.; Dempewolf, H.; Ramirez-Villegas, J.; Guarino, L.; Jarvis, A.; Rieseberg, L.H.; Struik, P.C. Increasing homogeneity in global food supplies and the implications for food security. Proc. Natl. Acad. Sci. USA 2014, 111, 4001–4006. [Google Scholar] [CrossRef] [PubMed]
- Tanksley, S.D.; McCouch, S.R. Seed banks and molecular maps: Unlocking genetic potential from the wild. Science 1997, 277, 1063–1066. [Google Scholar] [CrossRef] [PubMed]
- Dyer, G.A.; López-Feldman, A.; Yúnez-Naude, A.; Taylor, J.E. Genetic erosion in maize’s center of origin. Proc. Natl. Acad. Sci. USA 2014, 111, 14094–14099. [Google Scholar] [CrossRef] [PubMed]
- Maxted, N.; Kell, S.P. Establishment of a Global Network for the in Situ Conservation of Crop Wild Relatives: Status and Needs; FAO Commission on Genetic Resources for Food and Agriculture: Rome, Italy, 2009; p. 266. [Google Scholar]
- Maxted, N.; Ford-Lloyd, B.V.; Hawkes, J.G. Complementary conservation strategies. In Plant Genetic Conservation, The In Situ Approach; Maxted, N., Ford-Lloyd, B.V., Hawkes, J.G., Eds.; Springer: Dordrecht, The Netherlands, 1997; pp. 15–39. [Google Scholar]
- Gruber, K. Agrobiodiversity: The living library. Nature 2017, 544, S8–S10. [Google Scholar] [CrossRef] [PubMed]
- Koo, B.; Wright, B.D. The optimal timing of evaluation of genebank accessions and the effects of biotechnology. Am. J. Agric. Econ. 2000, 82, 797–811. [Google Scholar] [CrossRef]
- Brown, A.H.D. Core collections: A practical approach to genetic resources management. Genome 1989, 31, 818–824. [Google Scholar] [CrossRef]
- McKhann, H.I.; Camilleri, C.; Bérard, A.; Bataillon, T.; David, J.L.; Reboud, X.; Le Corre, V.; Caloustian, C.; Gut, I.G.; Brunel, D. Nested core collections maximizing genetic diversity in arabidopsis thaliana. Plant. J. 2004, 38, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.-W.; Chung, H.-K.; Cho, G.-T.; Ma, K.-H.; Chandrabalan, D.; Gwag, J.-G.; Kim, T.-S.; Cho, E.-G.; Park, Y.-J. Powercore: A program applying the advanced m strategy with a heuristic search for establishing core sets. Bioinformatics 2007, 23, 2155–2162. [Google Scholar] [CrossRef] [PubMed]
- Borrayo, E.; Takeya, M. Signal-Processing Tools for Core-Collection Selection from Genetic-Resource Collections. Available online: https://f1000research.com/articles/4-97/v1 (accessed on 14 September 2017).
- Khazaei, H.; Street, K.; Bari, A.; Mackay, M.; Stoddard, F.L. The figs (focused identification of germplasm strategy) approach identifies traits related to drought adaptation in vicia faba genetic resources. PLoS ONE 2013, 8, e63107. [Google Scholar] [CrossRef] [PubMed]
- Street, K.; Bari, A.; Mackay, M.; Amri, A. How the focused identification of germplasm strategy (figs) is used to mine plant genetic resources collections for adaptive traits. In Enhancing Crop Genepool Use: Capturing Wild Relative and Landrace Diversity for Crop Improvement; Maxted, N., Dulloo, M.E., Ford-Lloyd, B.V., Eds.; CABI: Boston, MA, USA, 2016; p. 54. [Google Scholar]
- Odong, T.L.; Jansen, J.; van Eeuwijk, F.A.; van Hintum, T.J.L. Quality of core collections for effective utilisation of genetic resources review, discussion and interpretation. Theor. Appl. Genet. 2013, 126, 289–305. [Google Scholar] [CrossRef] [PubMed]
- Fao: Second Global Plan of Action for Plant Genetic Resources. Available online: http://www.fao.org/agriculture/crops/thematic-sitemap/theme/seeds-pgr/gpa/en/ (accessed on 24 July 2017).
- Divseek. Available online: http://www.divseek.org (accessed on 21 July 2017).
- G2p-sol: Linking Genetic Resources, Genomes and Phenotypes of Solanaceous Crops. Available online: http://www.g2p-sol.eu/ (accessed on 21 January 2017).
- Traditom: Traditional Tomato Varieties and Cultural Practices. Available online: http://traditom.eu/ (accessed on 21 July 2017).
- Hajjar, R.; Hodgkin, T. The use of wild relatives in crop improvement: A survey of developments over the last 20 years. Euphytica 2007, 156, 1–13. [Google Scholar] [CrossRef]
- Harlan, J.R.; de Wet, J.M.J. Toward a rational classification of cultivated plants. Taxon 1971, 20, 509–517. [Google Scholar] [CrossRef]
- Gepts, P.; Papa, R. Possible effects of (trans)gene flow from crops on the genetic diversity from landraces and wild relatives. Environ. Biosaf. Res. 2003, 2, 89–103. [Google Scholar] [CrossRef]
- Cardi, T. Cisgenesis and genome editing: Combining concepts and efforts for a smarter use of genetic resources in crop breeding. Plant Breed. 2016, 135, 139–147. [Google Scholar] [CrossRef]
- Cardi, T.; D’Agostino, N.; Tripodi, P. Genetic transformation and genomic resources for next-generation precise genome engineering in vegetable crops. Front. Plant. Sci 2017, 8, 241. [Google Scholar] [CrossRef] [PubMed]
- Zamir, D. Improving plant breeding with exotic genetic libraries. Nat. Rev. Genet. 2001, 2, 983–989. [Google Scholar] [CrossRef] [PubMed]
- Prohens, J.; Gramazio, P.; Plazas, M.; Dempewolf, H.; Kilian, B.; Díez, M.J.; Fita, A.; Herraiz, F.J.; Rodríguez-Burruezo, A.; Soler, S.; et al. Introgressiomics: A new approach for using crop wild relatives in breeding for adaptation to climate change. Euphytica 2017, 213, 158. [Google Scholar] [CrossRef]
- Holland, J.B. Genetic architecture of complex traits in plants. Curr. Opin. Plant Biol. 2007, 10, 156–161. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Gore, M.; Buckler, E.S.; Yu, J. Status and prospects of association mapping in plants. Plant Genome 2008, 1, 5–20. [Google Scholar] [CrossRef]
- Korte, A.; Farlow, A. The advantages and limitations of trait analysis with gwas: A review. Plant Methods 2013, 9, 29. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.E.; Verbyla, K.L.; Verbyla, A.P.; Raghavan, C.; Singh, V.K.; Gaur, P.; Leung, H.; Varshney, R.K.; Cavanagh, C.R. Magic populations in crops: Current status and future prospects. Theor. Appl. Genet. 2015, 128, 999–1017. [Google Scholar] [CrossRef] [PubMed]
- King, E.G.; Merkes, C.M.; McNeil, C.L.; Hoofer, S.R.; Sen, S.; Broman, K.W.; Long, A.D.; Macdonald, S.J. Genetic dissection of a model complex trait using the drosophila synthetic population resource. Genome Res. 2012, 22, 1558–1566. [Google Scholar] [CrossRef] [PubMed]
- Pascual, L.; Desplat, N.; Huang, B.E.; Desgroux, A.; Bruguier, L.; Bouchet, J.-P.; Le, Q.H.; Chauchard, B.; Verschave, P.; Causse, M. Potential of a tomato magic population to decipher the genetic control of quantitative traits and detect causal variants in the resequencing era. Plant Biotechnol. J. 2015, 13, 565–577. [Google Scholar] [CrossRef] [PubMed]
- Alseekh, S.; Ofner, I.; Pleban, T.; Tripodi, P.; Di Dato, F.; Cammareri, M.; Mohammad, A.; Grandillo, S.; Fernie, A.R.; Zamir, D. Resolution by recombination: Breaking up solanum pennellii introgressions. Trends Plant Sci. 2013, 18, 536–538. [Google Scholar] [CrossRef] [PubMed]
- Henry, R.J. Plant Genotyping: The DNA Fingerprinting of Plants; CABI Publishing: Oxford, UK, 2001. [Google Scholar]
- Mammadov, J.; Aggarwal, R.; Buyyarapu, R.; Kumpatla, S. Snp markers and their impact on plant breeding. Int. J. Plant Genom. 2012, 2012, 728398. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.M.; Maughan, P.J. Snp genotyping using kaspar assays. In Plant genotyping: Methods and Protocols; Batley, J., Ed.; Springer: New York, NY, USA, 2015; pp. 243–256. [Google Scholar]
- Shen, G.-Q.; Abdullah, K.G.; Wang, Q.K. The taqman method for snp genotyping. In Single Nucleotide Polymorphisms: Methods and Protocols; Komar, A.A., Ed.; Humana Press: Totowa, NJ, USA, 2009; pp. 293–306. [Google Scholar]
- LaFramboise, T. Single nucleotide polymorphism arrays: A decade of biological, computational and technological advances. Nucleic Acids Res. 2009, 37, 4181–4193. [Google Scholar] [CrossRef] [PubMed]
- Sim, S.C.; Durstewitz, G.; Plieske, J.; Wieseke, R.; Ganal, M.W.; Van Deynze, A.; Hamilton, J.P.; Buell, C.R.; Causse, M.; Wijeratne, S.; et al. Development of a large snp genotyping array and generation of high-density genetic maps in tomato. PLoS ONE 2012, 7, e40563. [Google Scholar] [CrossRef] [PubMed]
- Jaccoud, D.; Peng, K.; Feinstein, D.; Kilian, A. Diversity arrays: A solid state technology for sequence information independent genotyping. Nucleic Acids Res. 2001, 29, E25. [Google Scholar] [CrossRef] [PubMed]
- Davey, J.W.; Hohenlohe, P.A.; Etter, P.D.; Boone, J.Q.; Catchen, J.M.; Blaxter, M.L. Genome-wide genetic marker discovery and genotyping using next-generation sequencing. Nat. Rev. Genet. 2011, 12, 499–510. [Google Scholar] [CrossRef] [PubMed]
- Ray, S.; Satya, P. Next generation sequencing technologies for next generation plant breeding. Front Plant Sci. 2014, 5, 367. [Google Scholar] [CrossRef] [PubMed]
- Aflitos, S.; Schijlen, E.; Jong, H.; Ridder, D.; Smit, S.; Finkers, R.; Wang, J.; Zhang, G.; Li, N.; Mao, L.; et al. Exploring genetic variation in the tomato (solanum section lycopersicon) clade by whole-genome sequencing. Plant. J. 2014, 80, 136–148. [Google Scholar] [PubMed]
- Schmutz, J.; McClean, P.E.; Mamidi, S.; Wu, G.A.; Cannon, S.B.; Grimwood, J.; Jenkins, J.; Shu, S.; Song, Q.; Chavarro, C.; et al. A reference genome for common bean and genome-wide analysis of dual domestications. Nat. Genet. 2014, 46, 707–713. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Zhang, J.; Sun, H.; Salse, J.; Lucas, W.J.; Zhang, H.; Zheng, Y.; Mao, L.; Ren, Y.; Wang, Z.; et al. The draft genome of watermelon (Citrullus lanatus) and resequencing of 20 diverse accessions. Nat. Genet. 2013, 45, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Causse, M.; Desplat, N.; Pascual, L.; Le Paslier, M.C.; Sauvage, C.; Bauchet, G.; Berard, A.; Bounon, R.; Tchoumakov, M.; Brunel, D.; et al. Whole genome resequencing in tomato reveals variation associated with introgression and breeding events. BMC Genom. 2013, 14, 791. [Google Scholar] [CrossRef] [PubMed]
- Terracciano, I.; Cantarella, C.; D’Agostino, N. Hybridization-based enrichment and next generation sequencing to explore genetic diversity in plants. In Dynamics of Mathematical Models in Biology: Bringing Mathematics to Life; Rogato, A., Zazzu, V., Guarracino, M., Eds.; Springer International Publishing: Cham, Switzerland, 2016; pp. 117–136. [Google Scholar]
- Terracciano, I.; Cantarella, C.; Fasano, C.; Cardi, T.; Mennella, G.; D’Agostino, N. Liquid-phase sequence capture and targeted re-sequencing revealed novel polymorphisms in tomato genes belonging to the mep carotenoid pathway. Sci. Rep. 2017, 7, 5616. [Google Scholar] [CrossRef] [PubMed]
- Ruggieri, V.; Anzar, I.; Paytuvi, A.; Calafiore, R.; Cigliano, R.A.; Sanseverino, W.; Barone, A. Exploiting the great potential of sequence capture data by a new tool, super-cap. DNA Res. 2017, 24, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Cronn, R.; Knaus, B.J.; Liston, A.; Maughan, P.J.; Parks, M.; Syring, J.V.; Udall, J. Targeted enrichment strategies for next-generation plant biology. Am. J. Bot. 2012, 99, 291–311. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Wang, H.; Michal, J.J.; Zhou, X.; Liu, B.; Woods, L.C.; Fuchs, R.A. Genome wide sampling sequencing for snp genotyping: Methods, challenges and future development. Int. J. Biol. Sci. 2016, 12, 100–108. [Google Scholar] [CrossRef] [PubMed]
- Baird, N.A.; Etter, P.D.; Atwood, T.S.; Currey, M.C.; Shiver, A.L.; Lewis, Z.A.; Selker, E.U.; Cresko, W.A.; Johnson, E.A. Rapid snp discovery and genetic mapping using sequenced rad markers. PLoS ONE 2008, 3, e3376. [Google Scholar] [CrossRef] [PubMed]
- Elshire, R.J.; Glaubitz, J.C.; Sun, Q.; Poland, J.A.; Kawamoto, K.; Buckler, E.S.; Mitchell, S.E. A robust, simple genotyping-by-sequencing (gbs) approach for high diversity species. PLoS ONE 2011, 6, e19379. [Google Scholar] [CrossRef] [PubMed]
- Voss-Fels, K.; Snowdon, R.J. Understanding and utilizing crop genome diversity via high-resolution genotyping. Plant. Biotechnol. J. 2016, 14, 1086–1094. [Google Scholar] [CrossRef] [PubMed]
- Taranto, F.; D’Agostino, N.; Tripodi, P. An overview of genotyping by sequencing in crop species and its application in pepper. In Dynamics of Mathematical Models in Biology: Bringing Mathematics to Life; Rogato, A., Zazzu, V., Guarracino, M., Eds.; Springer International Publishing: Cham, Switzerland, 2016; pp. 101–116. [Google Scholar]
- Boutet, G.; Alves Carvalho, S.; Falque, M.; Peterlongo, P.; Lhuillier, E.; Bouchez, O.; Lavaud, C.; Pilet-Nayel, M.-L.; Rivière, N.; Baranger, A. Snp discovery and genetic mapping using genotyping by sequencing of whole genome genomic DNA from a pea ril population. BMC Genom. 2016, 17, 121. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Gupta, S.; Bandhiwal, N.; Kumar, T.; Bharadwaj, C.; Bhatia, S. High-density linkage map construction and mapping of seed trait qtls in chickpea (Cicer arietinum L.) using genotyping-by-sequencing (gbs). Sci. Rep. 2015, 5, 17512. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; McGregor, C.; Zhang, Y.; Gong, G.; Zhang, H.; Guo, S.; Sun, H.; Cai, W.; Zhang, J.; Xu, Y. An integrated genetic map based on four mapping populations and quantitative trait loci associated with economically important traits in watermelon (Citrullus lanatus). BMC Plant. Biol. 2014, 14, 33. [Google Scholar] [CrossRef] [PubMed]
- Celik, I.; Gurbuz, N.; Uncu, A.T.; Frary, A.; Doganlar, S. Genome-wide snp discovery and qtl mapping for fruit quality traits in inbred backcross lines (ibls) of solanum pimpinellifolium using genotyping by sequencing. BMC Genom. 2017, 18, 1. [Google Scholar] [CrossRef] [PubMed]
- Sidhu, G.; Mohan, A.; Zheng, P.; Dhaliwal, A.K.; Main, D.; Gill, K.S. Sequencing-based high throughput mutation detection in bread wheat. BMC Genom. 2015, 16, 962. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.; Singh, A.; Sharma, M.; Kumar, P.; Roy, J. Development of ems-induced mutation population for amylose and resistant starch variation in bread wheat (Triticum aestivum) and identification of candidate genes responsible for amylose variation. BMC Plant Biol. 2016, 16, 217. [Google Scholar] [CrossRef] [PubMed]
- Taranto, F.; D’Agostino, N.; Greco, B.; Cardi, T.; Tripodi, P. Genome-wide snp discovery and population structure analysis in pepper (Capsicum annuum) using genotyping by sequencing. BMC Genom. 2016, 17, 943. [Google Scholar] [CrossRef] [PubMed]
- Pavan, S.; Lotti, C.; Marcotrigiano, A.R.; Mazzeo, R.; Bardaro, N.; Bracuto, V.; Ricciardi, F.; Taranto, F.; D’Agostino, N.; Schiavulli, A.; et al. A distinct genetic cluster in cultivated chickpea as revealed by genome-wide marker discovery and genotyping. Plant. Genome 2017, 10. [Google Scholar] [CrossRef] [PubMed]
- Poland, J.A.; Brown, P.J.; Sorrells, M.E.; Jannink, J.L. Development of high-density genetic maps for barley and wheat using a novel two-enzyme genotyping-by-sequencing approach. PLoS ONE 2012, 7, e32253. [Google Scholar] [CrossRef] [PubMed]
- Schröder, S. Optimization of genotyping by sequencing (gbs) data in common bean (Phaseolus vulgaris L.). Mol. Breed. 2016, 36, 6. [Google Scholar] [CrossRef]
- Torkamaneh, D.; Laroche, J.; Belzile, F. Genome-wide snp calling from genotyping by sequencing (gbs) data: A comparison of seven pipelines and two sequencing technologies. PLoS ONE 2016, 11, e0161333. [Google Scholar] [CrossRef] [PubMed]
- Catchen, J.; Hohenlohe, P.A.; Bassham, S.; Amores, A.; Cresko, W.A. Stacks: An analysis tool set for population genomics. Mol. Ecol. 2013, 22, 3124–3140. [Google Scholar] [CrossRef] [PubMed]
- Lu, F.; Lipka, A.E.; Glaubitz, J.; Elshire, R.; Cherney, J.H.; Casler, M.D.; Buckler, E.S.; Costich, D.E. Switchgrass genomic diversity, ploidy, and evolution: Novel insights from a network-based snp discovery protocol. PLoS Genet. 2013, 9, e1003215. [Google Scholar] [CrossRef] [PubMed]
- Russell, J.; Hackett, C.; Hedley, P.; Liu, H.; Milne, L.; Bayer, M.; Marshall, D.; Jorgensen, L.; Gordon, S.; Brennan, R. The use of genotyping by sequencing in blackcurrant (Ribes nigrum): Developing high-resolution linkage maps in species without reference genome sequences. Mol. Breed. 2014, 33, 835–849. [Google Scholar] [CrossRef]
- Huang, Y.F.; Poland, J.A.; Wight, C.P.; Jackson, E.W.; Tinker, N.A. Using genotyping-by-sequencing (gbs) for genomic discovery in cultivated oat. PLoS ONE 2014, 9, e102448. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [PubMed]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef] [PubMed]
- Awclust. Available online: https://sourceforge.net/projects/awclust/ (accessed on 20 July 2017).
- Yang, S.; Fresnedo-Ramirez, J.; Wang, M.; Cote, L.; Schweitzer, P.; Barba, P.; Takacs, E.M.; Clark, M.; Luby, J.; Manns, D.C.; et al. A next-generation marker genotyping platform (ampseq) in heterozygous crops: A case study for marker-assisted selection in grapevine. Hortic. Res. 2016, 3, 16002. [Google Scholar] [CrossRef] [PubMed]
- Buckler, E.S.; Ilut, D.C.; Wang, X.; Kretzschmar, T.; Gore, M.A.; Mitchell, S.E. Rampseq: Using repetitive sequences for robust genotyping. bioRxiv 2016. [Google Scholar] [CrossRef]
- Furbank, R.T.; Tester, M. Phenomics—Technologies to relieve the phenotyping bottleneck. Trends Plant Sci. 2011, 16, 635–644. [Google Scholar] [CrossRef] [PubMed]
- Araus, J.L.; Cairns, J.E. Field high-throughput phenotyping: The new crop breeding frontier. Trends Plant Sci. 2014, 19, 52–61. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zhang, Q.; Huang, D. A review of imaging techniques for plant phenotyping. Sensors 2014, 14, 20078–20111. [Google Scholar] [CrossRef] [PubMed]
- Fritsche-Neto, R.; Borém, A. Phenomics: How Next-Generation Phenotyping Is Revolutionizing Plant Breeding; Springer: Dordrecht, Switzerland, 2015; pp. 1–142. [Google Scholar]
- Liew, O.; Chong, P.; Li, B.; Asundi, A. Signature optical cues: Emerging technologies for monitoring plant health. Sensors 2008, 8, 3205–3239. [Google Scholar] [CrossRef] [PubMed]
- Mishra, K.B.; Iannacone, R.; Petrozza, A.; Mishra, A.; Armentano, N.; La Vecchia, G.; Trtílek, M.; Cellini, F.; Nedbal, L. Engineered drought tolerance in tomato plants is reflected in chlorophyll fluorescence emission. Plant Sci. 2012, 182, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Lootens, P.; Devacht, S.; Baert, J.; van Waes, J.; van Bockstaele, E.; Roldán-Ruiz, I. Evaluation of cold stress of young industrial chicory (Cichorium intybus L.) by chlorophyll a fluorescence imaging. Ii. Dark relaxation kinetics. Photosynthetica 2011, 49, 185–194. [Google Scholar] [CrossRef]
- Van der Heijden, G.; Song, Y.; Horgan, G.; Polder, G.; Dieleman, A.; Bink, M.; Palloix, A.; van Eeuwijk, F.; Glasbey, C. Spicy: Towards automated phenotyping of large pepper plants in the greenhouse. Funct. Plant Biol. 2012, 39, 870–877. [Google Scholar] [CrossRef]
- Rascher, U.; Blossfeld, S.; Fiorani, F.; Jahnke, S.; Jansen, M.; Kuhn, A.J.; Matsubara, S.; Märtin, L.L.A.; Merchant, A.; Metzner, R.; et al. Non-invasive approaches for phenotyping of enhanced performance traits in bean. Funct. Plant Biol. 2011, 38, 968–983. [Google Scholar] [CrossRef]
- Pauli, D.; Chapman, S.C.; Bart, R.; Topp, C.N.; Lawrence-Dill, C.J.; Poland, J.; Gore, M.A. The quest for understanding phenotypic variation via integrated approaches in the field environment. Plant Physiol. 2016, 172, 622–634. [Google Scholar] [CrossRef] [PubMed]
- White, J.W.; Andrade-Sanchez, P.; Gore, M.A.; Bronson, K.F.; Coffelt, T.A.; Conley, M.M.; Feldmann, K.A.; French, A.N.; Heun, J.T.; Hunsaker, D.J.; et al. Field-based phenomics for plant genetics research. Field Crop. Res. 2012, 133, 101–112. [Google Scholar] [CrossRef]
- Hirschhorn, J.N.; Daly, M.J. Genome-wide association studies for common diseases and complex traits. Nat. Rev. Genet. 2005, 6, 95–108. [Google Scholar] [CrossRef] [PubMed]
- Brachi, B.; Morris, G.P.; Borevitz, J.O. Genome-wide association studies in plants: The missing heritability is in the field. Genome Biol. 2011, 12, 232. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Han, B. Natural variations and genome-wide association studies in crop plants. Annu. Rev. Plant Biol. 2014, 65, 531–551. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhao, J.; Xu, Y.; Liang, J.; Chang, P.; Yan, F.; Li, M.; Liang, Y.; Zou, Z. Genome-wide association mapping for tomato volatiles positively contributing to tomato flavor. Front. Plant Sci. 2015, 6, 1042. [Google Scholar] [CrossRef] [PubMed]
- Nimmakayala, P.; Abburi, V.L.; Saminathan, T.; Alaparthi, S.B.; Almeida, A.; Davenport, B.; Nadimi, M.; Davidson, J.; Tonapi, K.; Yadav, L.; et al. Genome-wide diversity and association mapping for capsaicinoids and fruit weight in capsicum annuum L. Sci. Rep. 2016, 6, 38081. [Google Scholar] [CrossRef] [PubMed]
- Pavan, S.; Marcotrigiano, A.R.; Ciani, E.; Mazzeo, R.; Zonno, V.; Ruggieri, V.; Lotti, C.; Ricciardi, L. Genotyping-by-sequencing of a melon (Cucumis melo L.) germplasm collection from a secondary center of diversity highlights patterns of genetic variation and genomic features of different gene pools. BMC Genom. 2017, 18, 59. [Google Scholar] [CrossRef] [PubMed]
- Nimmakayala, P.; Tomason, Y.R.; Abburi, V.L.; Alvarado, A.; Saminathan, T.; Vajja, V.G.; Salazar, G.; Panicker, G.K.; Levi, A.; Wechter, W.P.; et al. Genome-wide differentiation of various melon horticultural groups for use in gwas for fruit firmness and construction of a high resolution genetic map. Front. Plant Sci. 2016, 7, 1437. [Google Scholar] [CrossRef] [PubMed]
- Cericola, F.; Portis, E.; Lanteri, S.; Toppino, L.; Barchi, L.; Acciarri, N.; Pulcini, L.; Sala, T.; Rotino, G.L. Linkage disequilibrium and genome-wide association analysis for anthocyanin pigmentation and fruit color in eggplant. BMC Genom. 2014, 15, 896. [Google Scholar] [CrossRef] [PubMed]
- Hart, J.P.; Griffiths, P.D. Genotyping-by-sequencing enabled mapping and marker development for the by-2 potyvirus resistance allele in common bean. Plant Genome 2015, 8. [Google Scholar] [CrossRef]
- Saxena, R.K.; Kale, S.M.; Kumar, V.; Parupali, S.; Joshi, S.; Singh, V.; Garg, V.; Das, R.R.; Sharma, M.; Yamini, K.N.; et al. Genotyping-by-sequencing of three mapping populations for identification of candidate genomic regions for resistance to sterility mosaic disease in pigeonpea. Sci. Rep. 2017, 7, 1813. [Google Scholar] [CrossRef] [PubMed]
- Bush, W.S.; Moore, J.H. Chapter 11: Genome-wide association studies. PLoS Comput. Biol. 2012, 8, e1002822. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Ersoz, E.; Lai, C.Q.; Todhunter, R.J.; Tiwari, H.K.; Gore, M.A.; Bradbury, P.J.; Yu, J.; Arnett, D.K.; Ordovas, J.M.; et al. Mixed linear model approach adapted for genome-wide association studies. Nat. Genet. 2010, 42, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Bhat, J.A.; Ali, S.; Salgotra, R.K.; Mir, Z.A.; Dutta, S.; Jadon, V.; Tyagi, A.; Mushtaq, M.; Jain, N.; Singh, P.K.; et al. Genomic selection in the era of next generation sequencing for complex traits in plant breeding. Front. Genet. 2016, 7, 221. [Google Scholar] [CrossRef] [PubMed]
- Hamblin, M.T.; Buckler, E.S.; Jannink, J.-L. Population genetics of genomics-based crop improvement methods. Trends Genet. 2011, 27, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Xie, C.; Wan, J.; He, Z.; Prasanna, B.M. Marker-assisted selection in cereals: Platforms, strategies and examples. In Cereal Genomics II; Gupta, P.K., Varshney, R.K., Eds.; Springer: Dordrecht, The Netherlands, 2013; pp. 375–411. [Google Scholar]
- He, S.; Schulthess, A.W.; Mirdita, V.; Zhao, Y.; Korzun, V.; Bothe, R.; Ebmeyer, E.; Reif, J.C.; Jiang, Y. Genomic selection in a commercial winter wheat population. Theor. Appl. Genet. 2016, 129, 641–651. [Google Scholar] [CrossRef] [PubMed]
- Duangjit, J.; Causse, M.; Sauvage, C. Efficiency of genomic selection for tomato fruit quality. Mol. Breed. 2016, 36, 29. [Google Scholar] [CrossRef]
- Yamamoto, E.; Matsunaga, H.; Onogi, A.; Ohyama, A.; Miyatake, K.; Yamaguchi, H.; Nunome, T.; Iwata, H.; Fukuoka, H. Efficiency of genomic selection for breeding population design and phenotype prediction in tomato. Heredity 2017, 118, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Druka, A.; Potokina, E.; Luo, Z.; Jiang, N.; Chen, X.; Kearsey, M.; Waugh, R. Expression quantitative trait loci analysis in plants. Plant Biotechnol. J. 2010, 8, 10–27. [Google Scholar] [CrossRef] [PubMed]
- Vidalis, A.; Živković, D.; Wardenaar, R.; Roquis, D.; Tellier, A.; Johannes, F. Methylome evolution in plants. Genome Biol. 2016, 17, 264. [Google Scholar] [CrossRef] [PubMed]
- Desta, Z.A.; Ortiz, R. Genomic selection: Genome-wide prediction in plant improvement. Trends Plant Sci 2014, 19, 592–601. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.-M.; Werner, D.J. Will the Traditional Horticultural Breeding and Genetics Research Be Fairly Valued in Academia. Available online: https://www.nature.com/articles/hortres201553 (accessed on 14 September 2017).



© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
D’Agostino, N.; Tripodi, P. NGS-Based Genotyping, High-Throughput Phenotyping and Genome-Wide Association Studies Laid the Foundations for Next-Generation Breeding in Horticultural Crops. Diversity 2017, 9, 38. https://doi.org/10.3390/d9030038
D’Agostino N, Tripodi P. NGS-Based Genotyping, High-Throughput Phenotyping and Genome-Wide Association Studies Laid the Foundations for Next-Generation Breeding in Horticultural Crops. Diversity. 2017; 9(3):38. https://doi.org/10.3390/d9030038
Chicago/Turabian StyleD’Agostino, Nunzio, and Pasquale Tripodi. 2017. "NGS-Based Genotyping, High-Throughput Phenotyping and Genome-Wide Association Studies Laid the Foundations for Next-Generation Breeding in Horticultural Crops" Diversity 9, no. 3: 38. https://doi.org/10.3390/d9030038
APA StyleD’Agostino, N., & Tripodi, P. (2017). NGS-Based Genotyping, High-Throughput Phenotyping and Genome-Wide Association Studies Laid the Foundations for Next-Generation Breeding in Horticultural Crops. Diversity, 9(3), 38. https://doi.org/10.3390/d9030038