Expressed Sequence Tag-Simple Sequence Repeat (EST-SSR) Marker Resources for Diversity Analysis of Mango (Mangifera indica L.)



Abstract
:
1. Introduction
2. Experimental Section
2.1. Plant Material

{kind=link}
{kind=link}
| Mangifera Variety | Species | Origin | Mangifera Variety | Species | Origin |
|---|---|---|---|---|---|
| Banana Callo | M. indica | Australia | Nam Doc Mai | M. indica | Thailand |
| Kensington Pride | M. indica | Australia | Irwin | M. indica | USA (Florida) |
| Alphonso | M. indica | India | Keitt | M. indica | USA (Florida) |
| Creeping | M. indica | India | Kent | M. indica | USA (Florida) |
| Hybrid 17 | M. indica | India | Lippens | M. indica | USA (Florida) |
| Neelum | M. indica | India | Palmer | M. indica | USA (Florida) |
| Padiri | M. indica | India | Tommy Atkins | M. indica | USA (Florida) |
| S.B. Chausa | M. indica | India | Van Dyke | M. indica | USA (Florida) |
| Suvarnarekha | M. indica | India | Sapa | M. indica (sens. let.) | Vietnam |
| Apple | M. indica | Malaysia | Xoài Cat Chu | M. indica | Vietnam |
| Arumanis | M. indica | Malesia | Julie | M. indica | West Indies |
| Tung Chi | M. indica (sens. let.) | Malesia | Binjai | M. caesia | Indonesia |
| Carabao Lamao | M. indica | Philippines | Bogor 2 | M. foetida | Indonesia |
| Willard | M. indica | Sri Lanka | Lomboc | M. laurina | Indonesia |
| Falan | M. indica | Thailand | Unknown | Mangifera sp. | Malaysia |
| Maha Chanook | M. indica | Thailand | Kweni | M. odorata | Malesia |
2.2. Phenotypic Evaluation of Mango Fruit
2.3. Genomic DNA Extraction
2.4. RNA Extraction
2.5. EST Library Construction, Sequencing and Annotation
2.6. EST Data Mining
| Variety | Tissue | Number of Reads | Average Length (nt) | Di | Tri | Tetra | Penta | Hexa | Total |
|---|---|---|---|---|---|---|---|---|---|
| Kensington Pride | Red Leaf | 6,304 | 473 | 84 | 347 | 12 | 3 | 8 | 454 |
| Kensington Pride | Fruit | 4,695 | 623 | 60 | 210 | 19 | 1 | 8 | 296 |
| Kensington Pride | Flower | 4,500 | 550 | 51 | 245 | 9 | 9 | 12 | 326 |
| Kensington Pride | Root | 5,302 | 704 | 39 | 355 | 22 | 2 | 20 | 438 |
| Irwin | Red Leaf | 4,039 | 564 | 62 | 210 | 8 | 4 | 2 | 286 |
| Total | 24,840 | 296 | 1,367 | 70 | 19 | 50 | 1,802 |
2.7. DNA Amplification and Capillary Electrophoresis
2.8. Data Analysis
3. Results
3.1. Analysis of Mango EST-SSR Sequences
3.2. Marker Development and Polymorphism of Mango EST-SSRs within Mangifera indica
3.3. Cross-Species Amplification
| Locus | GenBank Accession No. | Repeat Motif | Homology | e-value | Primer Sequence (5'-3') | M. indica | Mangifera Species | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Size Range | No. Alleles | HE | HO | PIC | Size Range | No. Alleles | ||||||
| QGMi001 | JZ532296 | (CCTTT)5 | Short vegetative phase
(controlling flowering time) | 4.00e − 51 | GAAAGGCTTGCAGAGACAGG | 171–227 | 7 | 0.690 | 0.667 | 0.633 | 171–228 | 6 |
| (floral development) | GTTTCTTCTGTTCGGTGATGGAGGAGT | |||||||||||
| QGMi002 | JZ532297 | (CTT)4 | Lacerata (CYP86A8) | 2.00e − 49 | GCTCAACCTCTTTCCTGCTC | 241–259 | 3 | 0.440 | 0.370 | 0.382 | 245–268 | 5 |
| (plant development) | GTTTCTTCAATCCCCAGAAGAAAACCA | |||||||||||
| QGMi003 | JZ532319 | (CTT)6 | TIR-NBS-LRR disease resistance gene | 3.00e − 24 | CAGGAATCTTCCCAAACGAA | 157–169 | 4 | 0.516 | 0.556 | 0.445 | 157–169 | 4 |
| (defence response) | GTTTCTTTGCCAGTGTCTTCACCTTCA | |||||||||||
| QGMi004 | JZ532302 | (AAG)5 | 9-cis epoxycarotenoid dioxygenase 5 | 2.00e − 44 | TTCACAACGAGAAGACATGGA | 236–244 | 7 | 0.784 | 0.593 | 0.732 | 233–245 | 7 |
| (abscisic acid biosynthesis; stress response) | GTTTCTTGGGACCTATTCGATCCCACT | |||||||||||
| QGMi005 | JZ532303 | (AAC)8 | WRKY40 | 2.00e − 53 | TGGAGGAATTGAACCGATTG | 303–318 | 6 | 0.752 | 0.519 | 0.691 | 303–324 | 4 |
| (transcription factor; defence response) | GTTTCTTCAGTATCGGAGGCGTCAGTC | |||||||||||
| QGMi006 | JZ532304 | (AAG)4 | Squalene monooxygenase | 7.00e − 58 | GCTTGCTTCGAGTTTTTGGT | 238 | 1 | ND | ND | ND | 238–241 | 2 |
| (isoprenoid biosynthesis) | GTTTCTTCGAGGAATGATCTCCGTTGT | |||||||||||
| QGMi007 | JZ532306 | (ATC)5 | KNAT1 (Brevipedicellus 1) | 3.00e − 37 | GCCTGAAGTAGTGGCTCGAC | 307–313 | 2 | 0.073 | 0.074 | 0.069 | 307 | 1 |
| (plant development) | GTTTCTTGAAATCCATGGCCTCCTGTA | |||||||||||
| QGMi008 | JZ532307 | (ATC)4 | WRKY7 | 9.00e − 13 | TCCAGCAATTTCCACCTTTC | 177 | 1 | ND | ND | ND | 177–179 | 2 |
| (transcription factor; stress response) | GTTTCTTTCACCATCACCAGTCAAGGA | |||||||||||
| QGMi009 | JZ532308 | (AT)29 | LRR transmembrane protein kinase | 1.00e + 00 | GGGTTAGCAAAACTGGTGGA | 156–228 | 13 | 0.872 | 0.556 | 0.843 | 156–212 | 4 |
| (cell signalling) | GTTTCTTCCCCAAGGATATACAGTAACCAG | |||||||||||
| QGMi010 | JZ532309 | (AGG)4 | Carotenoid cleavage dioxygenase 1 | 3.00e − 95 | GGTTTGAGCTTCCAAATTGC | 236–247 | 4 | 0.520 | 0.654 | 0.415 | 236–247 | 4 |
| (carotenoid biosynthesis) | GTTTCTTCCTGGGAAAGTCAACAGCAG | |||||||||||
| QGMi011 | JZ532312 | (CCGGCT)4 | Isopentenyl diphosphate isomerase 1 | 2.00e + 000 | CAACTTCCGAAAGCTAGAGGAG | 248–290 | 6 | 0.526 | 0.346 | 0.487 | 248–277 | 3 |
| (isoprenoid biosynthesis) | GTTTCTTCGTGGCACTCATTACCACAC | |||||||||||
| QGMi012 | JZ532313 | (AAG)5 | UDP glucosyltransferase | 4.00e − 77 | GGCTGAACTCAAAGGAACCA | 221–224 | 2 | 0.257 | 0.296 | 0.221 | 218–224 | 3 |
| (flavone biosynthesis) | GTTTCTTATAAGCCCTCTGCCTTCCAT | |||||||||||
| QGMi013 | JZ532314 | (AAG)6 | Ethylene responsive element binding factor 4 | 1.00e − 19 | ATCACGGTTCGGAGAGGTC | 200–206 | 3 | 0.423 | 0.519 | 0.375 | 197–206 | 3 |
| (transcription factor; stress response) | GTTTCTTGCAAAAACACGAGGACCAAT | |||||||||||
| QGMi014 | JZ532320 | (AAG)4 | Pectin methylesterase 3 | 9.00e − 78 | GCTTGCTTCGAGTTTTTGGT | 214–215 | 2 | 0.037 | 0.037 | 0.036 | 215–216 | 2 |
| (plant development; adventitious rooting) | GTTTCTTCGAGGAATGATCTCCGTTGT | |||||||||||
| QGMi015 | JZ532315 | (AAC)7 | KNAT3 (knotted1like homeobox gene 3) | 5.00e − 45 | CAACCACACTTCACGGACAC | 236–247 | 3 | 0.234 | 0.259 | 0.211 | 236–244 | 2 |
| (plant development) | GTTTCTTCATGTTTTCGCTGTTGCTGT | |||||||||||
| QGMi016 | JZ532316 | (ATCT)4 | Ultrapetala 1 | 6.00e − 52 | ACCAACGGCAACACCTACA | 257–266 | 4 | 0.666 | 0.667 | 0.585 | 251–258 | 4 |
| (plant development) | GTTTCTTCGGCAAATCAAAGGAAAGAA | |||||||||||
| QGMi017 | JZ532298 | (CTT)6 | Jasmonate insensitive 1 | 5.00e − 35 | GGAGAGAGTGCAGTGTCATGG | 110 | 1 | ND | ND | ND | 110 | 1 |
| (RNA transcription factor; stress response) | GTTTCTTATTGAAGGCGTTGTTGAAGC | |||||||||||
| QGMi018 | JZ532299 | (AATT)5 | MYB family transcription factor | 5.00e − 07 | GCTCTCTCTGTAACCTTCTTGTTT | 179–195 | 3 | 0.477 | 0.333 | 0.375 | 183–191 | 2 |
| (transcription factor) | GTTTCTTAGTTGTGTCCGTTGTTGCTG | |||||||||||
| QGMi019 | JZ532300 | (GCT)4 | Elongated hypocotyl 5 | 4.00e + 00 | CATGAAAAGAGATGAGGGAAA | 264 | 1 | ND | ND | ND | 262-264 | 2 |
| (plant development) | GTTTCTTCGGAGGCTCCAATGTAAAAC | |||||||||||
| QGMi020 | JZ532301 | (CT)7 | IAA-leucine resistant 3 | 2.00e − 51 | GCTCTGACGCGGAGATTC | 101–107 | 4 | 0.694 | 0.667 | 0.630 | 103–107 | 2 |
| (transcription factor) | GTTTCTTGTTGTTTTCCTGGCTGCAAT | |||||||||||
| QGMi021 | JZ532305 | (ATC)4 | WRKY DNA-binding protein 15 | 9.00e − 26 | GCAAGAACCAAGGTGGTGTT | 291 | 1 | ND | ND | ND | 291–294 | 2 |
| (transcription factor) | GTTTCTTCCGCTGAAGAAACCTGAGAC | |||||||||||
| QGMi022 | JZ532310 | (AAC)4 | MYB60 | 1.00e − 29 | CGTCTTCTCGAAGGATGGAT | 157 | 1 | ND | ND | ND | 154–157 | 2 |
| (transcription factor; stress response) | GTTTCTTCCTCCTTGTTTCTCCTCTTTCA | |||||||||||
| QGMi023 | JZ532311 | (AAC)7 | Phytochrome-associated protein 2 | 4.00e − 09 | TCAATGCAAAGAAGCTCTGAAA | 133–145 | 5 | 0.734 | 0.926 | 0.676 | 139–145 | 3 |
| (plant development) | GTTTCTTGCCTCAGCTCAGTCTCCTTG | |||||||||||
| QGMi024 | JZ532317 | (GATT)4 | MYB family transcription factor | 2.00e − 65 | CGCTTTCATCTGCTCAACTG | 245–249 | 3 | 0.237 | 0.111 | 0.217 | 246–250 | 3 |
| (transcription factor) | GTTTCTTACACCGCCGCAGCTC | |||||||||||
| QGMi025 | JZ532318 | (AGC)4 | WRKY DNA-binding protein 33 | 9.00e − 06 | TAGGGAAGCACAACCACGAT | 300–303 | 2 | 0.465 | 0.333 | 0.352 | 298–303 | 4 |
| (defence response) | GTTTCTTGTTCATCCTTGGCTCTCGAC | |||||||||||
| Locus | Unique Allele Size (bp) | Mangifera species |
|---|---|---|
| QGMi001 | 228 | Mangifera sp. |
| QGMi002 | 245*, 252#, 268^ | M. caesia*; Mangifera sp.#; M. foetida^ |
| QGMi004 | 233^, 245* | M. foetida^; M. caesia* |
| QGMi005 | 324 | M. caesia |
| QGMi006 | 241 | M. caesia |
| QGMi008 | 179 | M. caesia |
| QGMi009 | 212 | M. laurina |
| QGMi011 | 258 | Mangifera sp. |
| QGMi012 | 218 | M. caesia |
| QGMi013 | 197 | M. caesia |
| QGMi016 | 251 | M. caesia |
| QGMi019 | 262 | M. caesia |
| QGMi020 | nil | Failed to amplify in M. caesia |
| QGMi021 | 294 | M. caesia |
| QGMi022 | 154 | M. caesia |
| QGMi024 | nil | Failed to amplify in M. caesia |
| QGMi025 | 298 | M. foetida |
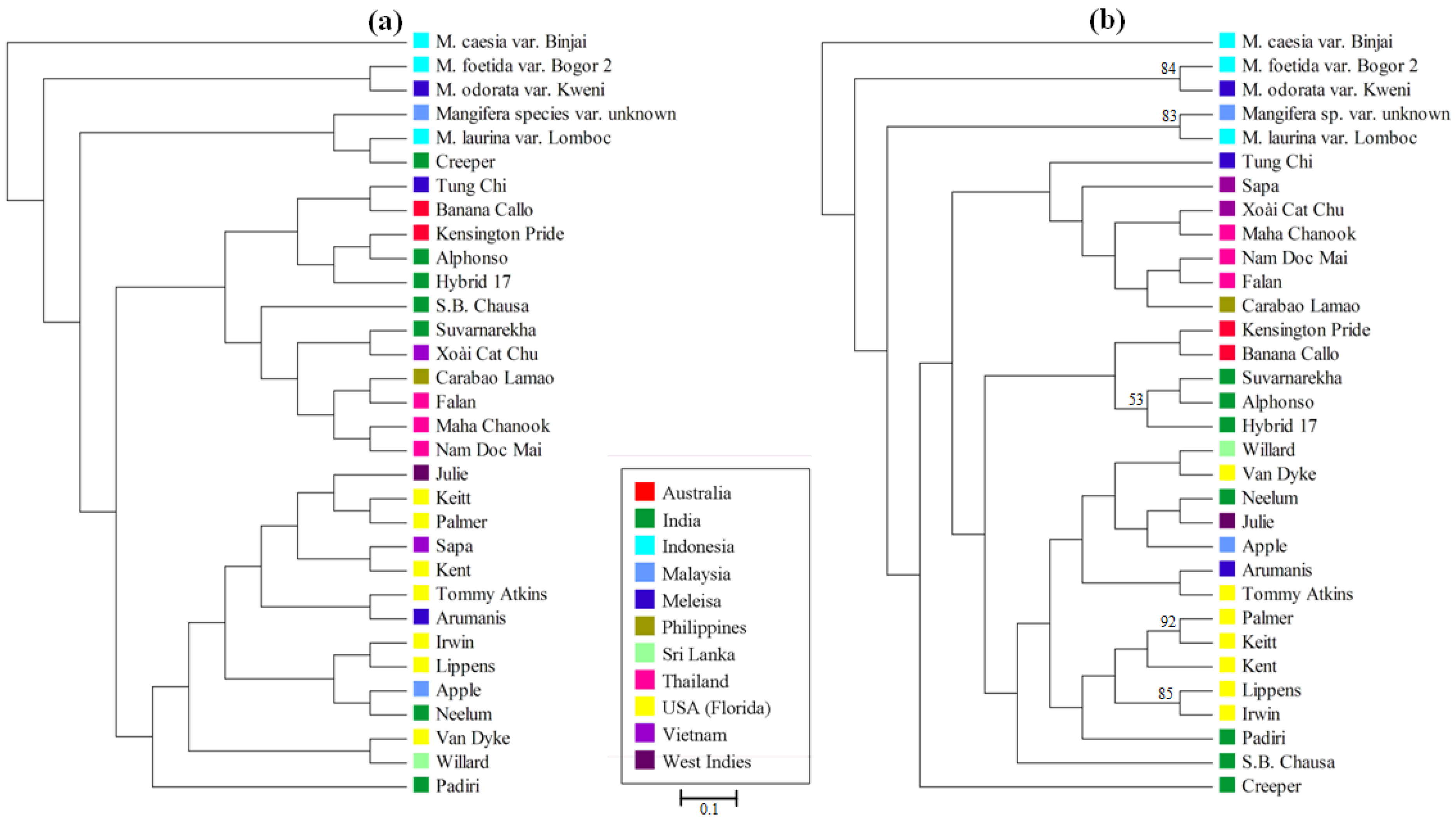
3.4. Mangifera Diversity Analysis
4. Discussion
5. Conclusions
Acknowledgments

Conflicts of Interest
References
- Kostermans, A.J.G.H.; Bompard, J.M. The Mangoes, Their Botany, Nomenclature, Horticulture and Utilisation; Academic Press: London, UK, 1993. [Google Scholar]
- FAOSTAT. Available online: http://faostat.fao.org/ (accessed on 15 November 2013).
- Stephens, S.E. Mango Varieties in Tropical Queensland; vol. 732, Queensland Department of Agriculture and Stock: Brisbane, Australia, 1963; pp. 1–4. [Google Scholar]
- Beal, P.R. New mango varieties. Qld. Agric. J. 1976, 120, 583–588. [Google Scholar]
- Catchpoole, D.; Bally, I.S.E. Search for Queensland’s top mango. Mango Care Newslett. 1990, 1, 6. [Google Scholar]
- Dillon, N.L.; Bally, I.S.E.; Wright, C.L.; Hucks, L.; Innes, D.J.; Dietzgen, R.G. Genetic diversity of the Australian National Mango Genebank. Scientia Hort. 2013, 150, 213–226. [Google Scholar] [CrossRef]
- Bally, I.S.E.; Lu, P.; Johnson, P.; Muller, W.J.; González, A. Past, Current and Future Approaches to Mango Genetic Improvement in Australia. In Proceedings of the 8th International Mango Symposium, Sun City, South Africa, 6–10 February 2006.
- Bally, I.S.E. Delta R2E2. New Mango for the Dry Tropics. HortNews, 31 October 1991; 12. [Google Scholar]
- Whiley, A.W. New Mango Variety Released. Mango Care Newslett. 2000, 29, 1. [Google Scholar]
- Holmes, R. Update on new mango varieties. Mango Care Newslett. 2002, 35, 10–11. [Google Scholar]
- Bally, I.S.E. New hybrids highlighted from National Mango Breeding Program. Mango Matters 2008, Summer, 8–14. [Google Scholar]
- Kashkush, K.; Jinggui, F.; Tomer, E.; Hillel, J.; Lavi, U. Cultivar identification and genetic map of mango (Mangifera indica). Euphytica 2001, 122, 129–136. [Google Scholar] [CrossRef]
- Chunwongse, J.; Phumichai, C.; Barbrasert, C.; Chunwongse, C.; Sukonsawan, S.; Boonreungrawd, R. Molecular mapping of mango cultivars “Alphonso” and “Palmar”. Acta Hortic. 2000, 509, 193–206. [Google Scholar]
- Gepts, P. Genetic markers and core collections. In Core Collections of Plant Genetic Resources; Hodgkin, T., Brown, A.H.D., van Hintum, T.J.L., Morales, E.A.V., Eds.; International Plant Genetic Institute (IPGRI)-John Wiley & Son: Chichester, UK, 1995; pp. 127–146. [Google Scholar]
- Duval, M.F.; Bunel, J.; Sitbon, C.; Risterucci, A.M.; Calabre, C.; Le Bellec, F. Genetic diversity of Caribbean mangoes (Mangifera indica L.) using microsatellite markers. Acta Hortic. 2006, 802, 183–188. [Google Scholar]
- Schnell, R.J.; Brown, J.S.; Olano, C.T.; Meerow, A.W.; Campbell, R.J.; Kuhn, D.N. Mango genetic diversity analysis and pedigree inferences for Florida cultivars using microsatellite markers. J. Am. Soc. Hortic. Sci. 2006, 131, 214–224. [Google Scholar]
- Ellis, J.R.; Burke, J.M. EST-SSRs as a resource for population genetic analyses. Heredity 2007, 99, 125–132. [Google Scholar] [CrossRef]
- Wöhrmann, T.; Weising, K. In silico mining for simple sequence repeat loci in pineapple expressed sequence tag database and cross-species amplification of EST-SSR markers across Bromeliaceae. Theor. Appl. Genet. 2011, 123, 635–647. [Google Scholar] [CrossRef]
- Huang, H.; Lu, J.; Ren, Z.; Hunter, W.; Dowd, S.E.; Dang, P. Minining and vaildating grape (Vitis. L.) ESTs to develop EST-SSR markers for genotyping and mapping. Mol. Breed. 2011, 28, 241–252. [Google Scholar]
- Hwang, J.H.; Ahn, S.G.; Oh, J.Y.; Choi, Y.W.; Kang, J.S.; Park, Y.H. Functional characterization of watermelon (Citrullus lanatus L.) EST-SSR by gel electrophoresis and high resolution melting analysis. Scientia Hort. 2011, 130, 715–724. [Google Scholar] [CrossRef]
- Pashley, C.H.; Ellis, J.R.; McCauley, D.E.; Burke, J.M. EST databases as a source for molecular markers: Lessons from Helianthus. J. Hered. 2006, 97, 381–388. [Google Scholar] [CrossRef]
- Chapman, M.A.; Hvala, J.; Strever, J.; Matvienko, M.; Kozik, A.; Michelmore, R.W.; Tang, S.; Knapp, S.J.; Burke, J.M. Development, polymorphism, and cross-taxon utility of EST-SSR markers from safflower (Carthamus tinctorius L.). Theor. Appl. Genet. 2009, 120, 85–91. [Google Scholar] [CrossRef]
- Varshney, R.K.; Graner, A.; Sorrells, M.E. Genic microsatellite markers in plants: Features and applications. Trends Biotechnol. 2005, 23, 48–55. [Google Scholar] [CrossRef]
- Chabane, K.; Ablett, G.; Cordeiro, G.; Valkoun, J.; Henry, R. EST versus genomic derived microsatellite markers for genotyping wild and cultivated barley. Genet. Res. Crop. Evol. 2005, 52, 903–909. [Google Scholar] [CrossRef]
- Kantety, R.V.; La Rota, M.; Matthews, D.E.; Sorrells, M.E. Data mining for simple sequence repeats in expressed sequence tags from barley, maize, rice, sorghum and wheat. Plant Mol. Biol. 2002, 48, 501–510. [Google Scholar] [CrossRef]
- Bandopadhyay, R.; Sharma, S.; Rustgi, S.; Singh, R.; Kumar, A.; Balyan, H.S.; Gupta, P.K. DNA polymorphism among 18 species of Triticum–Aegilops complex using wheat EST-SSRs. Plant Sci. 2004, 166, 349–356. [Google Scholar] [CrossRef]
- Fraser, L.G.; Harvey, C.F.; Crowhurst, R.N.; de Silva, H.N. EST-derived microsatellites from Actinidia species and their potential for mapping. Theor. Appl. Genet. 2004, 108, 1010–1016. [Google Scholar] [CrossRef]
- Depeiges, A.; Goubely, C.; Lenoir, A.; Cocherel, S.; Picard, G.; Raynal, M.; Grellet, F.; Delseny, M. Identification of the most represented repeated motifs in Arabidopsis thaliana microsatellite loci. Theor. Appl. Genet. 1995, 91, 160–168. [Google Scholar]
- Cordeiro, G.M.; Casu, R.; Mcintyre, C.L.; Manners, J.M.; Henry, R.J. Microsatellite markers from sugarcane Saccharum spp. ESTs cross transferable to Erianthus and sorghum. Plant Sci. 2001, 160, 1115–1123. [Google Scholar] [CrossRef]
- Lima, L.S.; Gramacho, K.P.; Gesteira, A.S.; Lopes, U.V.; Gaiotto, F.A.; Zaidan, H.A.; Pires, J.L.; Cascardo, J.C.M.; Micheli, F. Characterization of microsatellites from cacao-Moniliophthora perniciosa interaction expressed sequence tags. Mol. Breed. 2008, 22, 315–318. [Google Scholar] [CrossRef]
- De Keyser, E.; de Rick, J.; van Bockstaele, E. Discovery of species-wide EST-derived markers in Rhododendron by intron-flanking primer design. Mol. Breed. 2009, 23, 171–178. [Google Scholar] [CrossRef]
- Dietzgen, R.G.; Bally, I.S.E.; Devitt, L.C.; Dillon, N.L.; Fanning, K.; Gidley, M.; Holton, T.A.; Innes, D.J.; Karan, M.; Sheik-Jabbari, J.; et al. Mango Genetics Underpin Efficient Breeding for Variety Improvement. In Proceedings of the Seventh Australian Mango Conference, Cairns, Australia, 25–28 May 2009; pp. 10–12.
- Hunter, R.S. Minutes of the thirty-first meeting of the board of directors of the optical society of America, incorporated. J. Optical Soc. Amer. 1948, 38, 651. [Google Scholar]
- Ewing, B.; Green, P. Basecalling of automated sequencer traces using phred. II. Error probabilities. Genome Res. 1998, 8, 186–194. [Google Scholar]
- Ewing, B.; Hillier, L.; Wendl, M.; Green, P. Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res. 1998, 8, 175–185. [Google Scholar] [CrossRef]
- Staden, R.; Beal, K.F.; Bonfield, J.K. The Staden package, 1998. Methods Mol. Biol. 2000, 132, 115–130. [Google Scholar]
- Huang, X.; Madan, A. CAP3: A DNA sequence assembly program. Genome Res. 1999, 9, 868–877. [Google Scholar] [CrossRef]
- Swarbreck, D.; Wilks, C.; Lamesch, P.; Berardini, T.Z.; Garcia-Hernandez, M.; Foerster, H.; Li, D.; Meyer, T.; Muller, R.; Ploetz, L.; et al. The Arabidopsis Information Resource (TAIR): Gene structure and function annotation. Nucleic Acids Res. 2008, 36, D1009–D1014. [Google Scholar]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI_BLAST: A new generation of protein database programs. Nucleic Acids Res. 1997, 25, 22893402. [Google Scholar]
- Rozen, S.; Skaletsky, H. Primer3 on the WWW for general users and for biologist programmers. In Bioinformatics Methods and Protocols in the series Methods in Molecular Biology; Krawetz, S., Misener, S., Eds.; Humana Press: Totowa, NJ, USA, 2000; pp. 365–386. [Google Scholar]
- Cavalli-Sforza, L.L.; Edwards, A.W.F. Phylogenetic analysis: Models and estimation procedures. Am. J. Human Genet. 1967, 19, 233–257. [Google Scholar]
- Reynolds, J.; Weir, B.; Cockerham, C.C. Estimation of the coancestry coefficient: Basis for a short term genetic distance. Genetics 1983, 105, 767–779. [Google Scholar]
- Nei, M. Genetic distance between populations. Am. Nat. 1972, 106, 283–292. [Google Scholar]
- Nei, M.; Tajima, F.; Tateno, Y. Accuracy of estimated phylogenetic trees from molecular data. J. Mol. Evol. 1983, 19, 153–170. [Google Scholar] [CrossRef]
- Chapuis, M.P.; Estoup, A. Microsatellite null alleles and estimation of population differentiation. Mol. Biol. Evol. 2007, 24, 621–623. [Google Scholar] [CrossRef]
- Felsenstein, J. Phylogenies from gene frequencies: A statistical problem. Sys. Zool. 1985, 34, 300–311. [Google Scholar] [CrossRef]
- Saitou, N.; Nei, M. The neighbour-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar]
- Liu, K.; Muse, S.V. PowerMarker: Integrated analysis environment for genetic marker data. Bioinformatics 2005, 21, 2128–2129. [Google Scholar] [CrossRef]
- Kalinowski, S.T.; Taper, M.L.; Marshall, T.C. Revising how the computer program CERVUS accommodates genotyping error increases success in paternity assignment. Mol. Ecol. 2007, 16, 1099–1106. [Google Scholar] [CrossRef]
- Liu, B.H. Statistical Genomics. LINKAGE, Mapping and QTL Analysis; CRC Press: Boca Raton, FL, USA, 1998. [Google Scholar]
- Mantel, N. The detection of disease clustering and a generalized regression approach. Cancer Res. 1967, 27, 209–220. [Google Scholar]
- Polymorphic SSRs Mining for EST Data. Available online: http://www.bioinformatics.nl/tools/polyssr/ (accessed on 22 November 2013).
- Kumpatla, S.P.; Mukhopadhyay, S. Mining and survey of simple sequence repeats in expressed sequence tags of dicotyledonous species. Genome 2005, 48, 985–998. [Google Scholar] [CrossRef]
- Poncet, V.; Rondeau, M.; Tranchant, C.; Cayrel, A.; Hamon, S.; de Kochko, A.; Hamon, P. SSR mining in coffee tree EST databases: Potential use of EST-SSRs as markers for the Coffea genus. Mol. Gen. Genomics 2006, 276, 436–449. [Google Scholar] [CrossRef]
- Chen, C.; Zhou, P.; Choi, Y.A.; Huang, S.; Gmitter, F.G., Jr. Mining and characterizing microsatellites from citrus ESTs. Theor. Appl. Genet. 2006, 112, 1248–1257. [Google Scholar] [CrossRef]
- Metzgar, D.; Bytof, J.; Wills, C. Selection against frameshift mutations limits microsatellite expansion in coding DNA. Genome Res. 2000, 10, 72–80. [Google Scholar]
- Hou, D. Anacardiaceae, 4. Mangifera. In Flora Malesiana; Series I; vol. 8, van Steenis, C.G.G.J., Ed.; Rijksherbarium: Leiden, The Netherlands, 1978; pp. 395–440. [Google Scholar]
- Teo, L.L.; Kiew, R.; Set, O.; Lee, S.K.; Gan, Y.Y. Hybrid status of kuwini, Mangifera odorata (Anacardiaceae) verified by amplified fragment polymorphism. Mol. Ecol. 2002, 11, 1465–1469. [Google Scholar] [CrossRef]
- Kiew, R.; Teo, L.L.; Gan, Y.Y. Assessment of the hybrid status of some Malesian plants using Amplified Fragment Length Polymorphism. Telopea 2003, 10, 225–233. [Google Scholar]
- Yonemori, K.; Honsho, C.; Kanzaki, S.; Eiadthong, W.; Sugiura, A. Phylogenetic relationships of Mangifera species revealed by ITS sequences of nuclear ribosomal DNA and a possibility of their hybrid origin. Plant Syst. Evol. 2002, 231, 59–75. [Google Scholar] [CrossRef]
- Hidayat, T.; Pancoro, A.; Kusumawaty, D.; Eiadthong, W. Molecular diversification and phylogeny of Mangifera (Anacardiaceae) in Indonesia and Thailand. Int. J. Adv. Sci. Eng. Inf. Technol. 2011, 1, 88–91. [Google Scholar]
- Campbell, R.J. A Guide to Mangos in Florida, 1st ed.; Fairchild Tropical Garden: Miami, FL, USA, 1992. [Google Scholar]
- Olano, C.T.; Schnell, R.J.; Quintanilla, W.E.; Campbell, R.J. Pedigree analysis of Florida mango cultivars. Proc. Fla. State Hort. Soc. 2005, 118, 192–197. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Dillon, N.L.; Innes, D.J.; Bally, I.S.E.; Wright, C.L.; Devitt, L.C.; Dietzgen, R.G. Expressed Sequence Tag-Simple Sequence Repeat (EST-SSR) Marker Resources for Diversity Analysis of Mango (Mangifera indica L.). Diversity 2014, 6, 72-87. https://doi.org/10.3390/d6010072
Dillon NL, Innes DJ, Bally ISE, Wright CL, Devitt LC, Dietzgen RG. Expressed Sequence Tag-Simple Sequence Repeat (EST-SSR) Marker Resources for Diversity Analysis of Mango (Mangifera indica L.). Diversity. 2014; 6(1):72-87. https://doi.org/10.3390/d6010072
Chicago/Turabian StyleDillon, Natalie L., David J. Innes, Ian S. E. Bally, Carole L. Wright, Luke C. Devitt, and Ralf G. Dietzgen. 2014. "Expressed Sequence Tag-Simple Sequence Repeat (EST-SSR) Marker Resources for Diversity Analysis of Mango (Mangifera indica L.)" Diversity 6, no. 1: 72-87. https://doi.org/10.3390/d6010072
APA StyleDillon, N. L., Innes, D. J., Bally, I. S. E., Wright, C. L., Devitt, L. C., & Dietzgen, R. G. (2014). Expressed Sequence Tag-Simple Sequence Repeat (EST-SSR) Marker Resources for Diversity Analysis of Mango (Mangifera indica L.). Diversity, 6(1), 72-87. https://doi.org/10.3390/d6010072