1. Introduction
Population genetics describes a population that is not evolving as in Hardy–Weinberg equilibrium. A Hardy–Weinberg population must be of a very large population size, with individuals not isolated from each other, without net mutation, with no natural selection acting upon the population, and the mating must be random pairing [
1,
2]. In real populations, genetics predicts high dispersal rates and high levels of gene flow, preventing local populations from differentiating into new species. These populations approach the Hardy–Weinberg equilibrium state. Levins [
3] developed the first metapopulation model as a set of local populations connected by migrating individuals. The metapopulation concept is derived from the influence of area and isolation on the colonization and extinction of each subpopulation [
4]. Subpopulations usually inhabit an isolated habitat with patches of resources, and the degree of isolation may vary depending on the distance between patches. Metapopulation models consider each subpopulation as individual, and the dynamics of subpopulations are based on colonization-extinction equilibrium [
5,
6]. A metapopulation is composed of many small subpopulations. As described by the Hardy–Weinberg model, if the gene flow between subpopulations is small, they will move further away from the state of equilibrium, and these subpopulations will be highly isolated from each other.
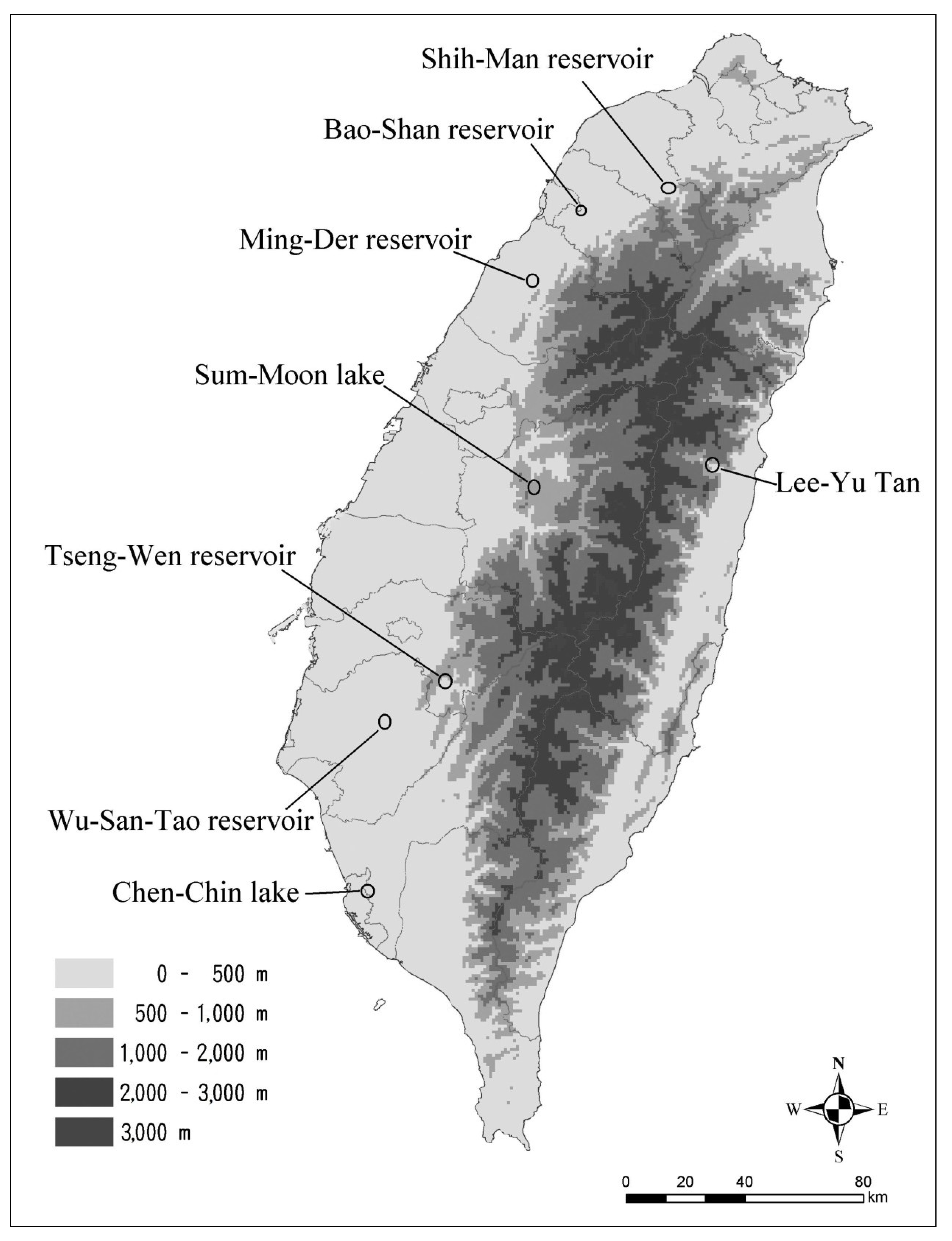
Freshwater invertebrates, including copepods, living in lakes or ponds and those whose habitats are not connected with each other by a waterway appear to be strong candidates for metapopulations [
7]. These freshwater bodies distributed on land are ideal analogs of oceanic islands, as per the first approach by MacArthur and Wilson [
4]. The dispersal ability of copepods may be dissimilar to that of other groups of animals with resting eggs or other dormancy mechanisms allowing for easy dispersal [
8]. These populations of copepods in reservoirs or lakes are isolated from each other to differing degrees owing to limited dispersal and low levels of gene flow, and the genetic structures of each population will be unique.
Knowledge of the early distribution pattern of Diaptomidae of Taiwan is limited. Kiefer [
9] described
Mongolodiaptomus formosanus (=
Mongolodiaptomus birulai (Rylov, 1922)) from Zaugatan (Sun-Moon Lake) in the third year after the reservoir was filled with water and, in another paper, also described this species in Wu-San-Tao reservoir [
10]. No reports of
Neodiaptomus schmackeri (Poppe and Richard, 1892) in Taiwan were made until it was first collected in Lee-Yu-Tan in 1996 by the author [
11]. After intense collection of samples, we found that most of the reservoir was dominated by
M. birulai, while in some places, both species coexisted, with minor populations of
N. schmackeri. At present, both species were the only freshwater calanoid copepods that could be found in the lowland reservoirs and fish ponds of Taiwan. Some species that had been recorded in the past became extinct after long-term environmental modification by modern agricultural development. Small animals in a freshwater ecosystem, such as zooplankton, at the microscopic scale are easier to ignore when discussing biodiversity decline. At present, more understanding of their metapopulation structure in an isolated habitat will be a crucial step for the conservation of plankton diversity. Isolated reservoirs around an island without water linking them are ideal sites to study the population genetics for small crustaceans.
In recent years, population genetics has employed molecular tools to study the genetic structure of different organisms. Genomic or mitochondrial DNA nucleotide sequences can explain the microevolution of populations of copepods [
12,
13,
14,
15,
16]. The aim of this study is to investigate the biodiversity of freshwater copepods at the genetic level. We used the mitochondria DNA cytochrome c oxidase subunit I (COI) gene as a marker to understand the genetic diversity and metapopulation structure of two freshwater copepods living in isolated water bodies around Taiwan.
4. Discussion
Based on the mtDNA COI gene sequence, Nei’s genetic distance between
N. schmackeri and
M. birulai is 0.23; this distance was smaller than that of some other groups of organisms between two different genera [
28,
29,
30]. Thrope and Sole-Cava [
30] suggested that a genetic distance of below 0.11 indicates conspecificity and one of above 0.22 corresponds to interspecific differentiation for invertebrates. For
N. schmackeri, the genetic distance between each population ranged from 0.013 to 0.058 and that for
M. birulai ranged from 0.004 to 0.016. Ferguson [
31] suggested that the use of genetic distance to infer separate species or several populations belonging to a single species is not parsimonious. The theoretical foundations are not well understood, and this method cannot be applied over a wide range of organisms.
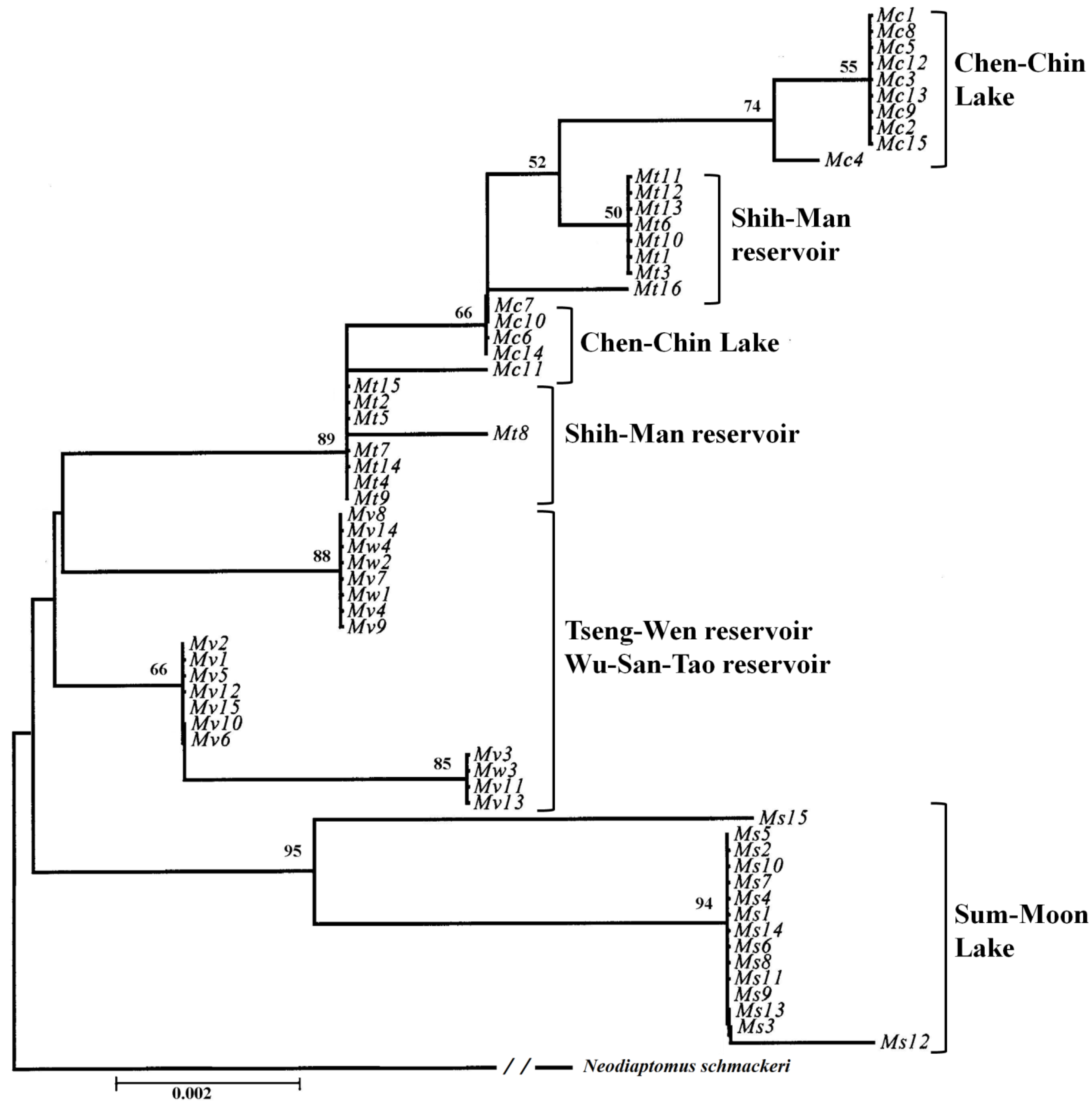
The nucleotide diversity (π) was calculated for all samples combined: π for N. schmackeri was 0.027, which was larger than that of M. birulai (0.008). We did not have N. schmackeri in Taiwan before 1940, based on the historical records. The higher diversity found in N. schmackeri may result by the founder effects of recent dispersion. At our sampling sites, if the dominant species was N. schmackeri, there was no M. birulai; conversely, if the dominant species was M. birulai, a minor population of N. schmackeri could coexist. The population genetic structure of M. birulai was more stable, with a larger population size, a wider distribution and a higher gene flow between subpopulations.
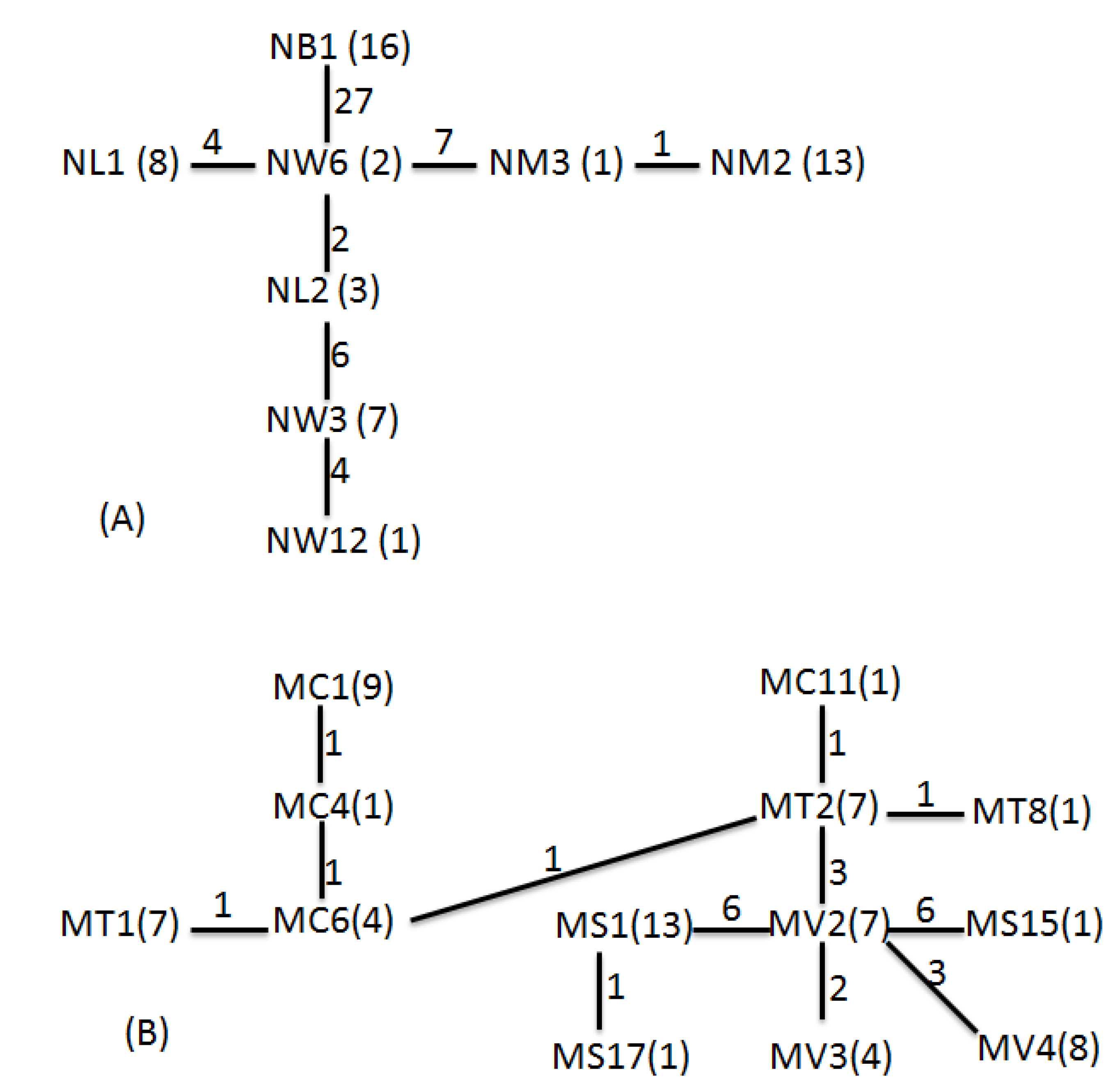
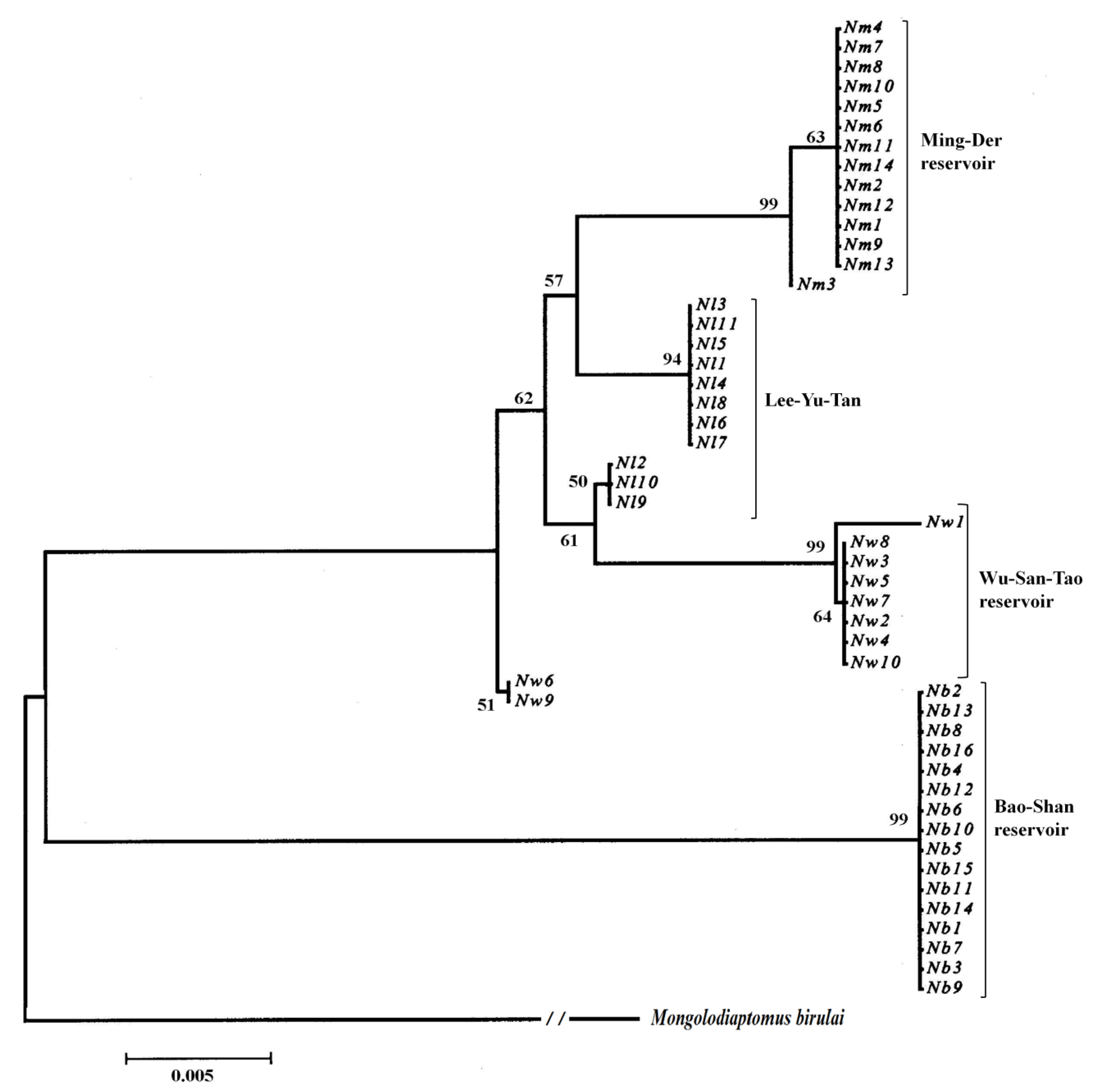
For N. schmackeri, Fst was 0.938 and Nm was 0.02 for all population pairs, and each population was isolated from the others, with little gene exchange. Relative to other reservoirs, Bao-Shan only had one haplotype and had the highest Fst and lowest Nm values (0.95–0.99; 0–0.01) compared with the other populations, isolated in recent years with no gene exchange. For M. birulai, Fst was 0.719 and Nm was 0.10 for all population pairs, and no population was seriously isolated from any of the others, as was the case for N. schmackeri. Sun-Moon Lake had the highest Fst (0.765–0.886) relative to the other populations. Tseng-Wen reservoir and Wu-San-Tao reservoir belong to the same river system and are linked by a canal; that could explain why they share most of the haplotypes and the greater gene flow and population differentiation with the lowest Fst (= 0.058) and highest Nm (= 4.40) values.
Freshwater zooplankton distributed in lakes or ponds are not always connected to each other, and the dispersal ability of zooplankton may vary in different groups of organisms. Recent studies have suggested that water birds could assist in the long-distance dispersal of rotifers, nematodes, mollusks, cladocerans and bryozoans [
8]. Most copepods live in inland waters, and dispersal may be mediated by flooding or locally-connected water channels [
32,
33,
34,
35]. Flooding can transport individuals to a wide range of lowland areas, and some can be buried in hyporheic sediment and secondarily emerge in surface water [
36]. Flooding is more common in lowland areas, and in reservoirs or highland lakes, dispersal and the establishment of populations may depend on river systems or dispersal methods other than flooding and carrying by birds. Therefore, these populations in reservoirs or lakes tend to be isolated from each other to differing degrees, owing to limited dispersal and low levels of gene flow, and the genetic structure of each population will be unique. Havel and Shurin [
37] suggested that the dispersion of freshwater zooplankton between wetlands with a limited distance gap (short distance; <10 km) is more rapid than a longer distance one. Relative to wetlands, dryer habitats with a gap longer than 10 km means that more isolated water bodies may constrain the geographical range and influence of the metapopulation structure. All of our sampling sites were further than 10 km apart and were isolated by different river systems, with the exception of Tseng-Wen reservoir and Wu-San-Tao reservoir.
Limited dispersal mechanisms may not wholly account for genetic divergence. De Meester
et al. [
38] proposed a monopolization hypothesis to explain a dispersal-gene flow paradox for freshwater invertebrates, that despite evidence of a high dispersal capacity, restricted gene flow is observed among multiple taxa. In the monopolization hypothesis, the early colonists develop such large populations that genetic contributions from later colonists are mathematically minor. De Meester
et al. [
38] also emphasize the importance of the role of local adaptation, highlighted by recent studies in zooplankton evolutionary ecology.
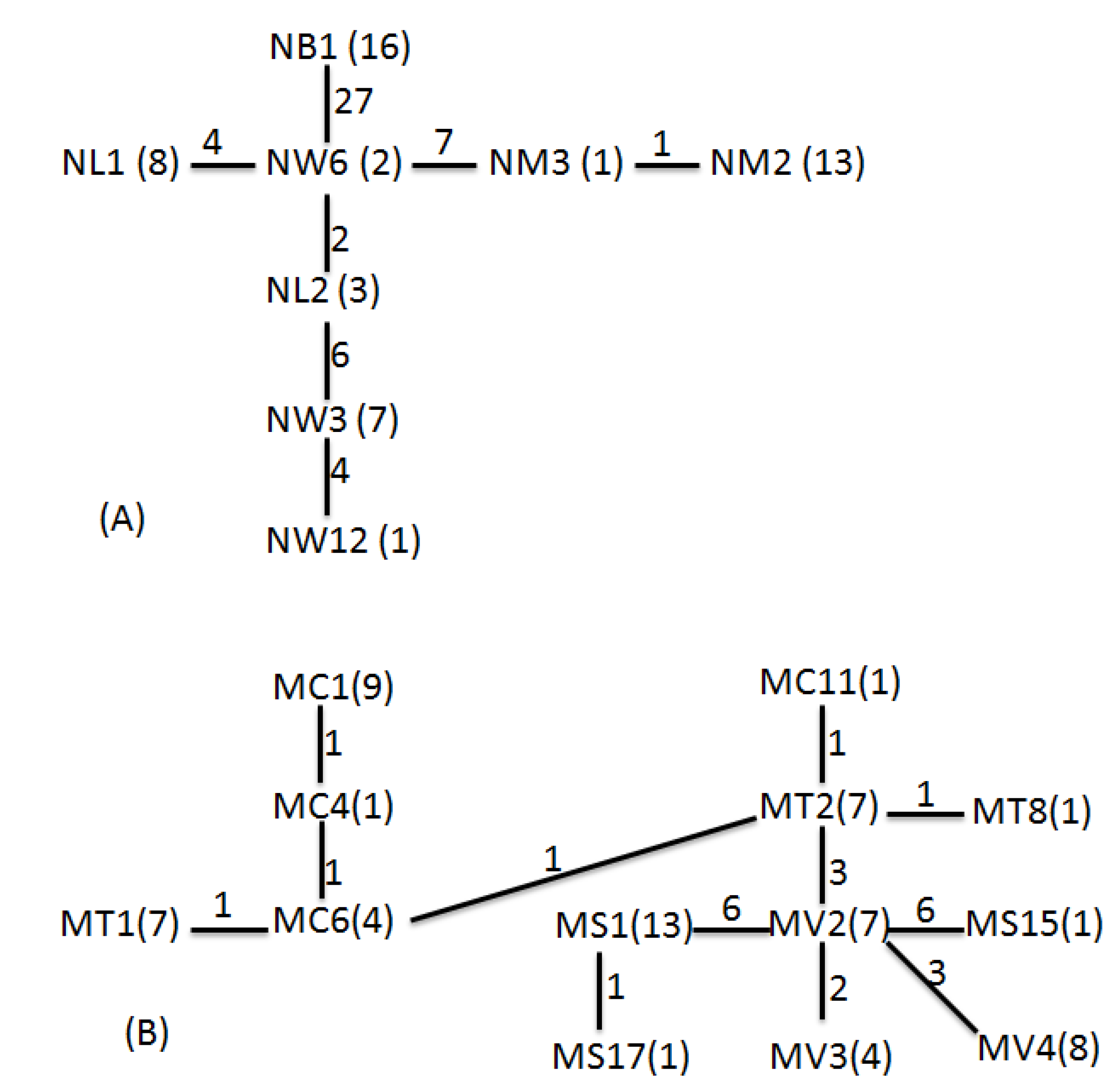
According the geophylogenetic trees and MSNs, the groupings of different sites were not based on geographical distance, as is usually the case; they were more or less constructed as according to the length of time for which the reservoir has been in use (
Table 7). Both species were centered on Wu-San-Tao reservoir (the oldest reservoir in Taiwan that is still functioning), and the terminal groups were the younger reservoirs, which came into use more recently. This could also explain how
N. schmackeri in the youngest of the reservoirs, Bao-Shan, had no haplotypic and nucleotidic diversity. The transporting of individuals to form new populations may depend on human activity, and a serial linage may be caused by the movement of construction machines containing water from reservoirs previously worked on. Another medium of transportation is the fishery industry at each reservoir: humans deliver living fish to the reservoir along with water, and some plankton may be transported to the new habitat in this way.
Table 7.
Collection site for each population and construction time of each reservoir.
Table 7.
Collection site for each population and construction time of each reservoir.
| Site of collection | Type | Time of construction | SP | Water source |
|---|
| Lee-Yu-Tan | lake | unknown | NS | Hualienchi |
| Wu-San-Tao | reservoir | 1930 | NS, MB | Tsengwenchi |
| Sum-Moon Lake | reservoir | 1934 | MB | Hsilochi |
| Chen-Chin Lake | reservoir | 1943 | MB | Kaopingchi |
| Shih-Man | reservoir | 1964 | NS, MB | Tahanchi |
| Ming-Der | reservoir | 1970 | NS | Houlungchi |
| Tseng-Wen | reservoir | 1973 | MB | Tsengwenchi |
| Bao-Shan | reservoir | 1985 | NS | Touchienchi |


{kind=link}
{kind=link}
{kind=link}
{kind=link}