1. Introduction
The sequencing and PCR amplification of 16S ribosomal RNA gene (16S rRNA) sequences have become the basis for the rapid identification and classification of Prokaryotes. Culture-independent methods take advantage of the robustness of the highly conserved 16S rRNA gene for phylogenetic assignments within microbial communities from any environment. The presence of this highly conserved gene in all Prokaryotes enables it to be used as a molecular chronometer in evolutionary studies and its length is ideal for all sequencing platforms [
1]. Next generation sequencing produces many thousands of small 16S rRNA reads per sample [
2,
3,
4,
5] that require a further classification step using specific 16S rRNA databases. Projects such as the Ribosomal Database Project II (RDP-II) at Michigan State University [
6,
7] and the Greengenes Project, maintained by the Lawrence Berkeley National Laboratory [
8] offer access to sets of rRNA sequence databases useful in microbial population studies. Those databases can be downloaded directly from the database project websites and can be tailored to the interests of the user such as the selection of the sequences in the database from cultured and/or uncultured organisms. However, these databases do not include all of the taxonomic information that is available for these sequences in their headers.
To address this problem we created TaxCollector (Taxonomy Collector), a set of Python scripts that attach taxonomic information from domain to species levels acquired from NCBI to RDP-II and Greengenes 16S rRNA databases. Thus, TaxCollector creates modified RDP or Greengenes databases with all of the taxonomic information to allow the user to rapidly identify taxonomic differences between communities at all levels. In addition to providing the code, up-to-date TaxCollector-modified 16S rRNA databases are available for download at
htp://www.microgator.org/. The TaxCollector code can also be quickly adapted to any other gene with an available database.
2. Experimental Section
2.1. Sampling, DNA Extraction and Sequencing
Two independent pyrosequencing-generated, 16S rRNA fragment libraries were used to demonstrate TaxCollector. The first set of sequences contained barcoded sequences amplified from DNA isolated from a sugar cane field in the Everglades Agricultural Area in Florida, first published by Roesch
et al. [
2]. Details on the sampling, DNA extraction, PCR amplification and sequencing for this library were described previously [
2] with the exceptions that only one round of PCR was done with primers containing the pyrosequencing 454 A and B adaptors and 454 FLX sequencing was done which resulted in an average read length of 223 bases.
The second set of sequences was obtained from the 16S rRNA amplification products of DNA isolated from fresh stool samples obtained from seven children at the Shands Hospital, University of Florida, Gainesville, FL. Samples were collected in sterile flask containers and kept at −20 ºC for further DNA extraction. DNA isolation and purification, PCR amplification and sequencing were performed as described by Roesch
et al. [
9]. After sequencing, pre-processing of the 454-sequences datasets was performed using to remove short sequences and trim those sequences that contain bases with low quality scores using PANGEA [
10].
2.4. Concatenation of the RDP and Greengenes Databases
After processed using TaxCollector, RDP and Greengenes databases were concatenated using a Python script called
remdup.py. Upon encountering records with duplicate (2 or more) headers, the script keeps the one that contains the largest sequence (
Table 1).
2.5. Utility of the TaxCollector modified databases using two datasets
The sequences were taxonomically classified using standalone BLAST search using Megablast. Megablast is a program inserted into the package called BLAST [
11] available in the NCBI website [
http://www.ncbi.nlm.nih.gov/blast/download.shtml]. Local blast analysis was performed against the five databases generated by TaxCollector (
Table 2). The closest bacterial relatives were assigned according to their best matches to sequences in the database.
Table 2.
Number of 16S rRNA sequences in six databases obtained before and after TaxCollector.
Table 2.
Number of 16S rRNA sequences in six databases obtained before and after TaxCollector.
| Database | Number of sequences |
|---|
| Downloaded | After TaxCollector | % of recovery sequences |
|---|
| RDP (isolates only) | 167,313 | 164,476 | 98.30 |
| RDP | 924,043 | 919,524 | 99.51 |
| Greengenes (isolates only) | 302,066 | 83,263* | 27.56 |
| Greengenes | 302,066 | 302,066 | 100.00 |
| RDP + Greengenes (isolates only) | 247,739 | 165,107 | 66.65 |
| RDP (Bacteria + Archaea, isolates only) | 169,386 | 166,543 | 98.32 |
To get an overall comparison and to determine differences in the number of classifiable sequences from each database, queries were grouped into Operational Taxonomic Units (OTU) based on the relatedness of sequences (
Table 3). In this study, queries/subjects were grouped into OTU exhibiting similarity values depending on the desired taxonomic level,
i.e., 80% at Domain/Phylum, 90% to Class/Order/Family, 95% to Genus (or corresponding OTU) and 99% of similarity to Species (or corresponding OTU) levels [
12]. The output file generated by Megablast was processed using PANGEA [
10].
Table 3.
Number of OTU identified in each taxonomic level, using six different databases modified by TaxCollector compared to the results obtained from the online RDP Classifier.
Table 3.
Number of OTU identified in each taxonomic level, using six different databases modified by TaxCollector compared to the results obtained from the online RDP Classifier.
| Database | Number OTU identified in each taxonomic level |
| Domain (80%) | Phylum (80%) | Class (90%) | Order (90%) | Family (90%) | Genus (95%) |
| RDP | | | | | | |
| | human gut | 1 | 4 | 5 | 11 | 17 | 25 |
| | Florida soil | 1 | 18 | 20 | 33 | 50 | 60 |
| RDP (isolates only) | | | | | | |
| | human gut | 1 | 4 | 5 | 12 | 21 | 31 |
| | Florida soil | 1 | 21 | 33 | 64 | 155 | 180 |
| Greengenes | | | | | | |
| | human gut | 1 | 4 | 6 | 9 | 14 | 26 |
| | Florida soil | 1 | 19 | 21 | 36 | 61 | 69 |
| Greengenes (isolates only) | | | | | | |
| | human gut | 1 | 4 | 5 | 11 | 20 | 30 |
| | Florida soil | 1 | 22 | 36 | 66 | 157 | 191 |
RDP + Greengenes
(isolates only) | | | | | | |
| human gut | 1 | 4 | 5 | 11 | 20 | 34 |
| Florida soil | 1 | 22 | 37 | 66 | 147 | 180 |
RDP Bac/Arch
(isolates only) | | | | | | |
| Florida soil | 2 | 22 | 37 | 70 | 158 | 184 |
| RDP Classifier | | | | | | |
| human gut | 1 | 4 | 5 | 11 | 20 | 19 |
| Florida soil | 2 | 13 | 15 | 15 | 16 | 17 |
2.6. RDP Classifier
The datasets were submitted to the RDP Classifier program of the Ribosomal Database Project (RDP-II) release 10 [
http://rdp.cme.msu.edu] to obtain the closest matches to known organisms using 16S rRNA gene fragments. Taxonomic hierarchy was performed on the Phylum, Class, Order, Family and Genus levels. Sequences were clustered at 80% of similarity for Phylum, 90% of similarity for Class/Order/Family and 95% of similarity for Genus [
11]. Tables were generated containing the percentage of each taxonomic level in each sample using text editors.
2.7. Megan
Reads were assigned using Megan, a metagenome package, which classifies DNA fragments based on a lowest common ancestor algorithm [
13]. Prior to the Megan analysis, the sequences were classified using the standalone BLAST search [
11]. Local blast analysis was performed against the RDP database containing sequences belonging to the isolates only. The BLAST output was processed by Megan to assess the taxonomy for each sequence. Results were exported and then placed into a text editor.
3. Results
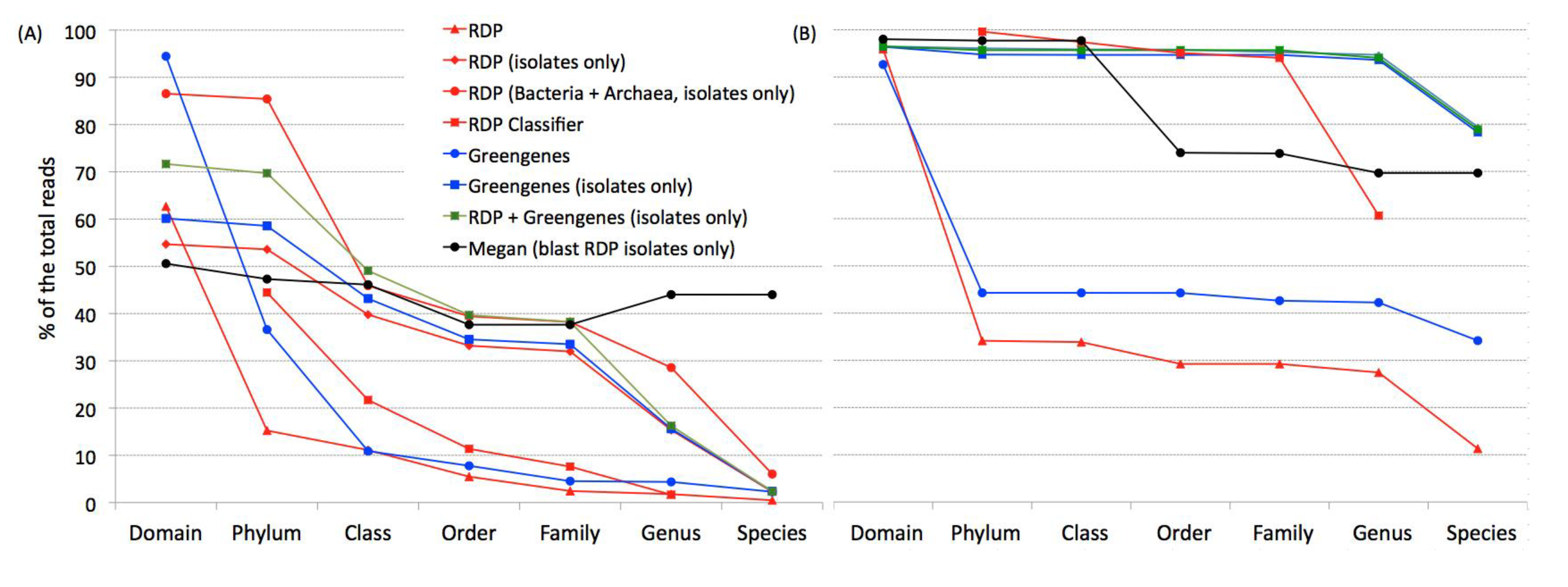
The databases modified by TaxCollector were tested using two 454 pyrosequencing libraries derived from two very different sources: human stool and soil sample. The identification of these sequences from the TaxCollector-modified databases was compared with the classification provided by RDP Classifier and Megan. A total of 8,006 16S rRNA sequences were analyzed for each sample.
The databases used for classification consisted of variants of the RDP-II and Greengenes databases modified to contain the taxonomic information in the header of each sequence's published name (
Figure 1). In addition, the total number of sequences available from RDP-II and Greengenes for this work and the number of sequences obtained after TaxCollector processing are listed (
Table 2). After processing using TaxCollector, a small proportion of sequences was not classified and thus is not inserted in the new database.
Figure 1.
Example of RDP-II database modified by TaxCollector. Top: a single 16S rRNA sequence downloaded directly from RDP-II. Bottom: the same sequence modified using TaxCollector.
Figure 1.
Example of RDP-II database modified by TaxCollector. Top: a single 16S rRNA sequence downloaded directly from RDP-II. Bottom: the same sequence modified using TaxCollector.
The sequences from the stool sample contain only bacterial sequences since the primers used for the amplification of 16S rRNA are bacterial-specific. In contrast, the soil sample sequences were generated using a primer set that amplifies 16S rRNA genes from both archaeal and bacterial groups.
As the Greengenes and RDP-II databases are both widely used, TaxCollector was used to make a set of modified databases that are specific to either Greengenes or RDP-II as well as additional databases that contain sequences from both sources. RDP-II allows the user to download sequences derived from only cultured isolates or from uncultured strains as well. The Greengenes databases include all Archaea and Bacteria while sequences from both domains are downloaded separately from the RDP-II databases.
Automated monthly updates of ten TaxCollector modified databases are available for download at
http://www.microgator.org/. These databases include all of the combinations described above.
When classifying the sequences from the two datasets used here, a far greater proportion of sequences are classified when the database includes only isolates (
Figure 2) since a large proportion of sequences are most closely related to a sequence from an uncultured organism. However, these uncultured sequences mask the identity of closely related organisms at traditional taxonomic levels.
These data also show that there is no value added to the taxonomic identification of the sequences when using databases from Greengenes compared to RDP-II. This is expected as the RDP-II database has more sequences than the Greengenes database. Tremendous added value was obtained for the soil sample when the archaeal sequences were included with the bacterial sequences in the RDP-II isolates database.
A far greater proportion of the human samples were classified at all taxonomic levels compared to the soil sample. As the RDP Classifier [
14] also uses sequences from uncultured organisms, it was unable to classify nearly as many sequences as the RDP databases designed to include sequences only from cultured organisms. Hence, RDP Classifier identified far fewer sequences at all taxonomic levels with the soil sample and at the genus level for the human sample. The same was observed in the classification using Megan. Both RDP Classifier and Megan classified more sequences from clinical samples than from soil sample.
Figure 2.
Microbial classifications obtained from modified RDP and Greengenes databases. Percentage of classifiable sequences in soil (A) and human (B) samples using six databases-header modified by TaxCollector, RDP Classifier and Megan.
Figure 2.
Microbial classifications obtained from modified RDP and Greengenes databases. Percentage of classifiable sequences in soil (A) and human (B) samples using six databases-header modified by TaxCollector, RDP Classifier and Megan.
4. Discussion
The 16S rRNA gene continues to play a very valuable role in the identification of Prokaryotes even with the increasing use of metagenomic sequencing for environmental and clinical samples [
15]. The advent of culture-independent techniques allows obtaining a large number of DNA sequences directly from environment samples. Tools such as BLAST, a similarity-based binning method, allow the classification of thousands of 16S rRNA reads using databases such as RDP-II and Greengenes databases [
11]. Depending on the database chosen by the user, the closest match for each sequence may be a cultured or uncultured organism but often these matches do not include useful taxonomic information. TaxCollector was designed to attach self-explanatory taxonomic information to RDP-II and Greengenes databases thus providing the ability to classify reads from domain to species level, thus providing self-explanatory taxonomic headers in the 16S rRNA gene databases. This is a significant advantage over the currently available databases where taxonomic information above the genus level is lacking.
Current tools for the classification of 16S rRNA do not provide all phylogenetic levels from domain to species directly in the database sequences’ headers. The tools provided within TaxCollector allows the creation of an up-to-date 16S rRNA database that ties NCBI taxonomic information with rRNA sequences from the Ribosomal Database Project (
http://rdp.cme.msu.edu) and/or Greengenes (
http://greengenes.lbl.gov). The code provided with TaxCollector is freely available and can be used for the creation of databases for other genes or sets of genes. The code can also be used to construct user-defined 16S rRNA databases.
The taxonomic information for the construction of TaxCollector databases is derived from NCBI and includes files called names and nodes. The taxonomic information in these files is highly curated by the NCBI Taxonomy team and is based on information from The Prokaryotes (
http://www.springerlink.com/reference-works/?sortorder=asc&mode=boolean&k=ti:(prokaryotes)), Bergey's manual (
http://www.cme.msu.edu/bergeys/), the up-to-date DSMZ Bacterial Nomenclature list (
http://www.dsmz.de/bactnom/bactname.htm), the Greengenes 16S rRNA database and workbench (
http://greengenes.lbl.gov), the International journal of Systematic and Evolutionary Microbiology (
http://ijs.sgmjournals.org/), the Official List of Bacterial Names with Standing in Nomenclature (
http://www.bacterio.cict.fr/), the Ribosomal Database Project (
http://rdp.cme.msu.edu), and the Taxonomic Outline of Bacteria and Archaea (
http://www.taxonomicoutline.org/). In addition, the NCBI taxonomic files are commonly used as the backbone of bacterial and archaeal taxonomic information for several international resources such as Megan [
13], the PhyloGenie [
16], and the Biopathway Workbench [
17].
Here TaxCollector was used to classify 16S rRNA pyrosequences from two very different environments: human stool and soil samples. As the soil dataset has many more OTU than the stool dataset, a much higher percentage of the sequences were identified to genus and species in the stool samples (
Figure 2). The data presented here illustrate that soil bacteria are far more diverse than gut bacteria (
Table 2). Five times more phyla are observed in the soil environment than in the human gut. Hence, the soil sample is phylum rich and species rich while the human sample is phylum poor and species rich.
Other tools can provide taxonomic information for 16S rRNA sequences but they are specifically designed for metagenome studies such as Megan [
13] and Sort-ITEMS [
18]. Using the TaxCollector modified database in PANGEA [
10], the classification of each sequence occurs during the MEGABLAST analysis and prior to clustering. In Megan, the BLAST analysis and classification are separate events requiring more processing time and more computer power.



{kind=link}
{kind=link}