



Phylogeographic Analyses of the Viviparous Multiocellated Racerunner (Eremias multiocellata) in the Tarim Basin of China


Abstract
1. Introduction
2. Materials and Methods
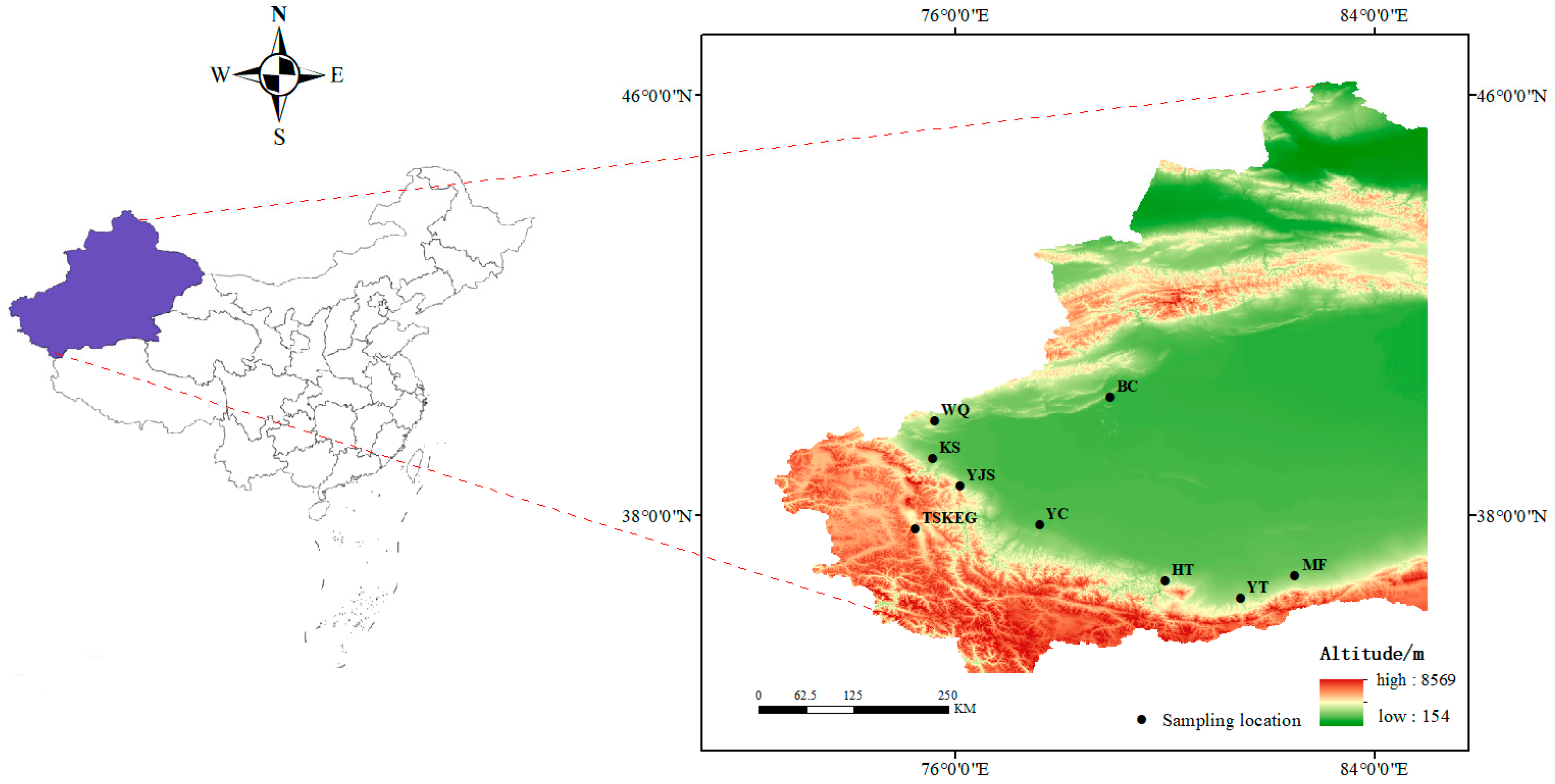
2.1. Sample Collection
2.2. mtDNA Gene Sequencing
2.3. Phylogenetic Analyses
2.4. Divergence Time Estimation
3. Results
3.1. mtDNA Gene Sequencing Results
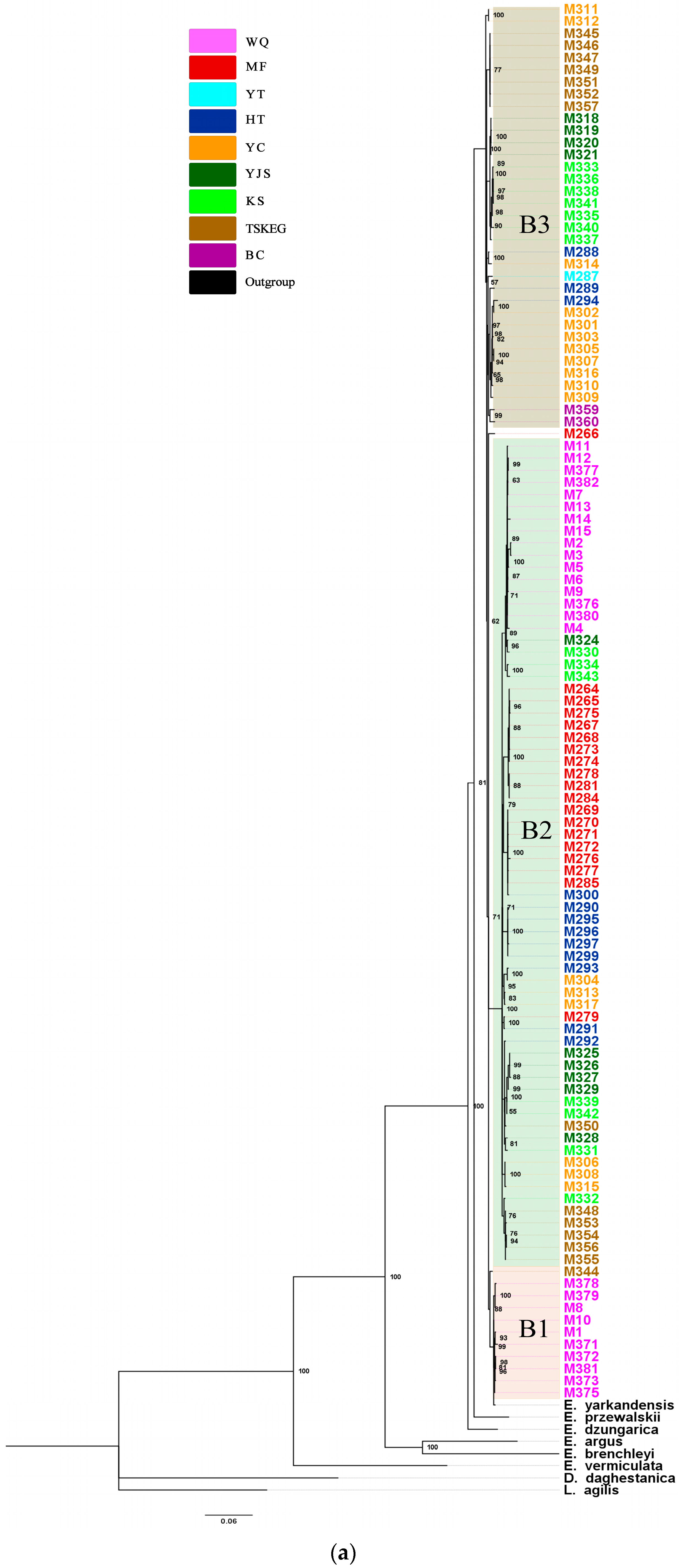
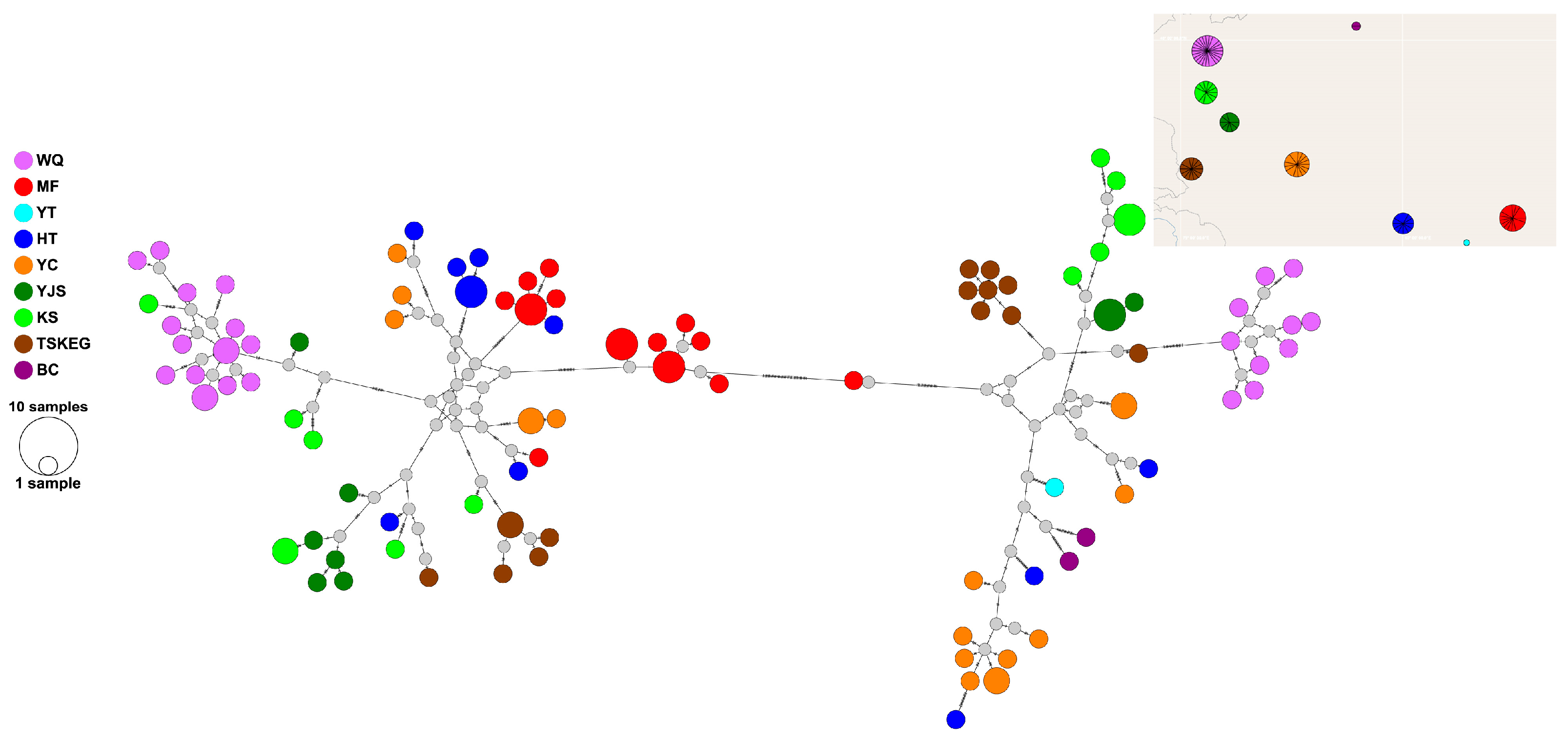
3.2. Phylogenetic Analysis Results
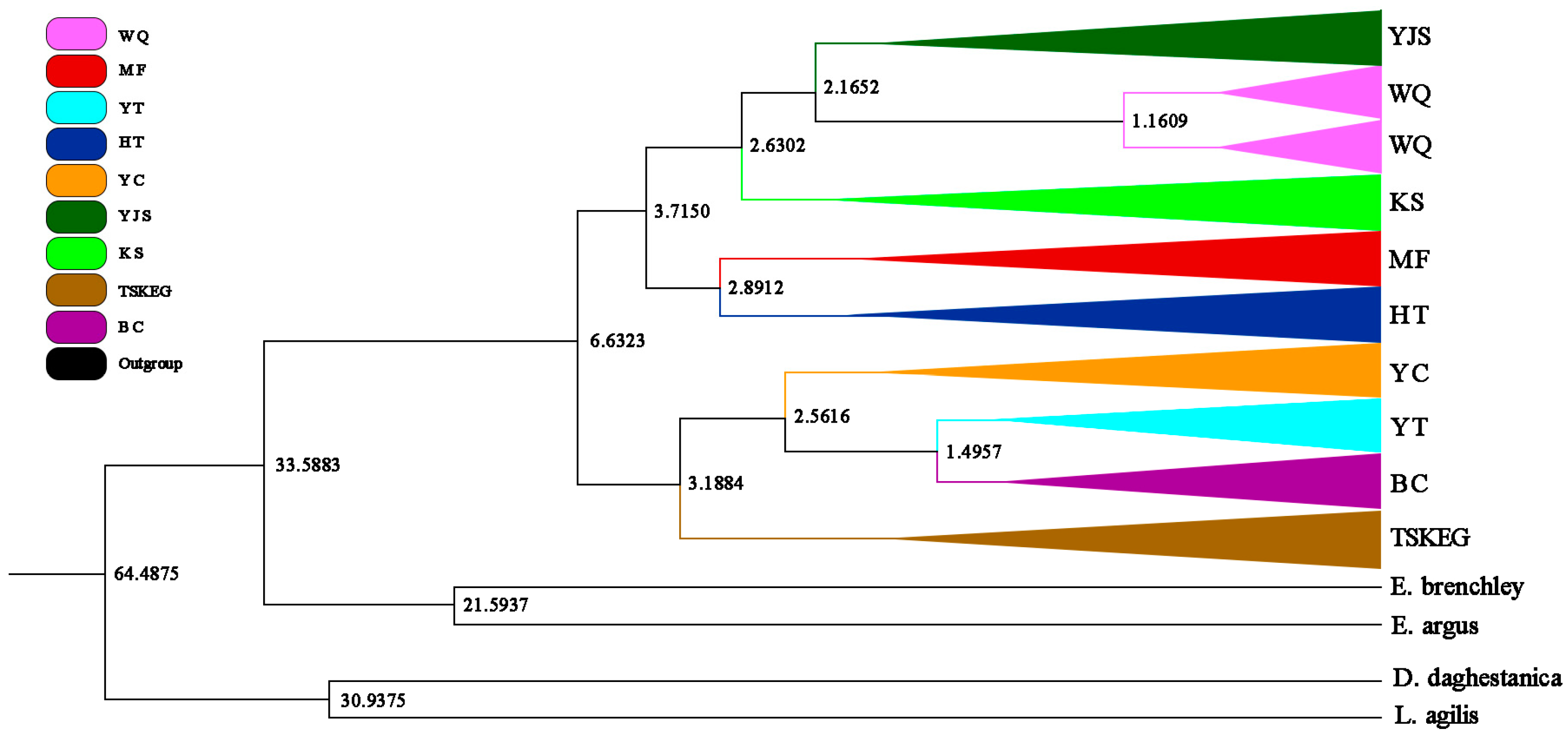
3.3. Divergence Time Estimation Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, Z.; Han, W.; Zhang, H.; Lou, Y.; Liu, X. Late Miocene-Pleistocene aridification of Asian inland revealed by sediments at Boguzihe section from western Tarim Basin, NW China. J. Arid. Land Resour. Environ. 2017, 31, 144–151. [Google Scholar]
- Meng, H.-H.; Gao, X.-Y.; Huang, J.-F.; Zhang, M.-L. Plant phylogeography in arid Northwest China: Retrospectives and perspectives. J. Syst. Evol. 2015, 53, 33–46. [Google Scholar] [CrossRef]
- Xu, Z.; Zhang, M.-L. Phylogeography of the Arid Shrub Atraphaxis frutescens (Polygonaceae) in Northwestern China: Evidence From cpDNA Sequences. J. Hered. 2015, 106, 184–195. [Google Scholar] [CrossRef]
- Guo, Z.T.; Ruddiman, W.F.; Hao, Q.Z.; Wu, H.B.; Qiao, Y.S.; Zhu, R.X.; Peng, S.Z.; Wei, J.J.; Yuan, B.Y.; Liu, T.S. Onset of Asian desertification by 22 Myr ago inferred from loess deposits in China. Nature 2002, 416, 159–163. [Google Scholar] [CrossRef]
- Sun, J.; Zhang, L.; Deng, C.; Zhu, R. Evidence for enhanced aridity in the Tarim Basin of China since 5.3 Ma. Quat. Sci. Rev. 2008, 27, 1012–1023. [Google Scholar] [CrossRef]
- Sun, J.; Zhang, Z.; Zhang, L. New evidence on the age of the Taklimakan Desert. Geology 2009, 37, 159–162. [Google Scholar] [CrossRef]
- Sun, J.M.; Liu, T.S. The age of the Taklimakan Desert. Science 2006, 312, 1621. [Google Scholar] [CrossRef]
- An, Z.S.; Kutzbach, J.E.; Prell, W.L.; Porter, S.C. Evolution of Asian monsoons and phased uplift of the Himalayan Tibetan plateau since Late Miocene times. Nature 2001, 411, 62–66. [Google Scholar] [CrossRef]
- Lu, H.; Yi, S.; Liu, Z.; Mason, J.A.; Jiang, D.; Cheng, J.; Stevens, T.; Xu, Z.; Zhang, E.; Jin, L.; et al. Variation of East Asian monsoon precipitation during the past 21 k.y. and potential CO2 forcing. Geology 2013, 41, 1023–1026. [Google Scholar] [CrossRef]
- Miao, Y.; Herrmann, M.; Wu, F.; Yan, X.; Yang, S. What controlled Mid-Late Miocene long-term aridification in Central Asia?—Global cooling or Tibetan Plateau uplift: A review. Earth-Sci. Rev. 2012, 112, 155–172. [Google Scholar] [CrossRef]
- Yang, X.P.; Rost, K.T.; Lehmkuhl, F.; Zhenda, Z.; Dodson, J. The evolution of dry lands in northern China and in the Republic of Mongolia since the Last Glacial Maximum. Quat. Int. 2004, 118, 69–85. [Google Scholar] [CrossRef]
- Yang, X.P.; Zhu, Z.D.; Jaekel, D.; Owen, L.A.; Han, J.M. Late Quaternary palaeoenvironment change and landscape evolution along the Keriya River, Xinjiang, China: The relationship between high mountain glaciation and landscape evolution in foreland desert regions. Quat. Int. 2002, 97–98, 155–166. [Google Scholar] [CrossRef]
- Feng, Q.; Su, Z.Z.; Jin, H.J. Desert evolution and climatic changes in the Tarim River basin since 12 ka BP. Sci. China Ser. D-Earth Sci. 1999, 42, 101–112. [Google Scholar] [CrossRef]
- Zhang, H.; Wu, H.W.; Zheng, Q.H.; Yu, Y.H. A preliminary study of oasis evolution in the Tarim Basin, Xinjiang, China. J. Arid. Environ. 2003, 55, 545–553. [Google Scholar] [CrossRef]
- Wang, W.; Lu, A.-M.; Ren, Y.; Endress, M.E.; Chen, Z.-D. Phylogeny and classification of Ranunculales: Evidence from four molecular loci and morphological data. Perspect. Plant Ecol. Evol. Syst. 2009, 11, 81–110. [Google Scholar] [CrossRef]
- Cho, Y.H.; Seo, C.W.; Jung, P.E.; Lim, Y.W. Global phylogeographical distribution of Gloeoporus dichrous. PLoS ONE 2023, 18, e0288498. [Google Scholar] [CrossRef]
- Li, W.Q.; Li, Y.; Wang, X.W.; Zhao, G.Q.; Zhu, G.R.; Cao, K.; Fang, W.C.; Wu, J.L.; Ma, K.; Chen, C.W.; et al. Genomic analysis provides insights into the westward expansion of domesticated peaches in China. Hortic. Plant J. 2024, 10, 367–375. [Google Scholar] [CrossRef]
- Su, Z.; Richardson, B.A.; Zhuo, L.; Jiang, X.; Li, W.; Kang, X. Genetic diversity and structure of an endangered desert shrub and the implications for conservation. AoB PLANTS 2017, 9, plx016. [Google Scholar] [CrossRef]
- Su, Z.; Wang, L.; Zhuo, L.; Jiang, X.; Li, W. Genetic diversity patterns in an endangered desert shrub from north-western China: Implications for conservation. Aust. J. Bot. 2019, 67, 571–584. [Google Scholar] [CrossRef]
- Zhang, H.-X.; Wang, Q.; Jia, S.-W. Genomic Phylogeography of Gymnocarpos przewalskii (Caryophyllaceae): Insights into Habitat Fragmentation in Arid Northwestern China. Diversity 2020, 12, 335. [Google Scholar] [CrossRef]
- Jia, S.-W.; Zhang, M.-L. Introgression of phylogeography lineages of Convolvulus gortschakovii (Convolvulaceae) in the northwest China. Plant Syst. Evol. 2021, 307, 19. [Google Scholar] [CrossRef]
- Yisilam, G.; Wang, C.-X.; Xia, M.-Q.; Comes, H.P.; Li, P.; Li, J.; Tian, X.-M. Phylogeography and Population Genetics Analyses Reveal Evolutionary History of the Desert Resource Plant Lycium ruthenicum (Solanaceae). Front. Plant Sci. 2022, 13, 915526. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Ma, S.; Sun, F.; Wei, B.; Nie, Y. Spatial genetic patterns of the medicinal and edible shrub Lycium ruthenicum (Solanaceae) in arid Xinjiang, China. Tree Genet. Genomes 2021, 17, 22. [Google Scholar] [CrossRef]
- Shi, X.; Jiang, X.; Zhang, H.; Qiu, J. Phylogeography and Genetic Structure of Sand Dune Specialist Stilpnolepis centiflora (Asteraceae) in Northwest China Revealed by Molecular Data. Diversity 2022, 14, 104. [Google Scholar] [CrossRef]
- Ababaikeri, B.; Zhang, Y.; Dai, H.; Shan, W. Revealing the coexistence of differentiation and communication in an endemic hare, Lepus yarkandensis (Mammalia, Leporidae) using specific-length amplified fragment sequencing. Front. Zool. 2021, 18, 50. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.H.; Xia, L.; Zhang, Q.; Yang, Q.S. Habitat fragmentation affects genetic diversity and differentiation of the Yarkand hare. Conserv. Genet. 2010, 11, 183–194. [Google Scholar] [CrossRef]
- Shan, W.J.; Tursun, M.; Zhou, S.Y.; Zhang, Y.C.; Dai, H.Y. Complete mitochondrial genome sequence of Gunther, 1875 (Lagomorpha, Leporidae): Characterization and phylogenetic analysis. Zookeys 2021, 1012, 135–149. [Google Scholar] [CrossRef]
- Abduriyim, S. Population Genetics of Gazella Subgotturosa in Xinjiang Based on the Mitochondrial DNA Cytochrome b Gene. Master’s Thesis, Xinjiang University, Ürümqi, China, 2015. (In Chinese). [Google Scholar]
- Hu, P.; Shao, Y.; Xu, J.; Wang, T.; Li, Y.; Liu, H.; Rong, M.; Su, W.; Chen, B.; Cui, S.; et al. Genome-wide study on genetic diversity and phylogeny of five species in the genus Cervus. BMC Genom. 2019, 20, 384. [Google Scholar] [CrossRef]
- Lorenzini, R.; Garofalo, L. Insights into the evolutionary history of Cervus (Cervidae, tribe Cervini) based on Bayesian analysis of mitochondrial marker sequences, with first indications for a new species. J. Zool. Syst. Evol. Res. 2015, 53, 340–349. [Google Scholar] [CrossRef]
- Mahmut, H.; Masuda, R.; Onuma, M.; Takahashi, M.; Nagata, J.; Suzuki, M.; Ohtaishi, N. Molecular phylogeography of the red deer (Cervus elaphus) populations in Xinjiang of China: Comparison with other Asian, European, and North American populations. Zool. Sci. 2002, 19, 485–495. [Google Scholar] [CrossRef]
- Araya-Donoso, R.; San Juan, E.; Tamburrino, I.; Lamborot, M.; Veloso, C.; Véliz, D. Integrating genetics, physiology and morphology to study desert adaptation in a lizard species. J. Anim. Ecol. 2022, 91, 1148–1162. [Google Scholar] [CrossRef]
- Yuan, X. Species diversity, threat factors and protection countermeasures of amphibians and reptiles in Tarim Populus euphratica National Nature Reserve. For. Xinjiang 2023, 3, 42–44+48. (In Chinese) [Google Scholar]
- Jiang, Z.G.; Wang, Y.Z.; Zhang, E.; Zhang, Y.Y.; Li, L.L.; Xie, F.; Cai, B.; Cao, L.; Zheng, G.M.; Dong, L.; et al. Red List of China’s Vertebrates. Biodivers. Sci. 2016, 24, 500–551. [Google Scholar]
- He, J.X.; Yan, C.; Holyoak, M.; Wan, X.R.; Ren, G.Y.; Hou, Y.F.; Xie, Y.; Zhang, Z.B. Quantifying the effects of climate and anthropogenic change on regional species loss in China. PLoS ONE 2018, 13, e0199735. [Google Scholar] [CrossRef]
- Wang, Y.Z.; Cai, B.; Li, J.T. China’s Red List of Biodiversity·Vertebrates (Vol. 3): Reptilies; Science Press: Beijing, China, 2021; pp. 43–49. [Google Scholar]
- Cox, N.; Young, B.E.; Bowles, P.; Fernandez, M.; Marin, J.; Rapacciuolo, G.; Böhm, M.; Brooks, T.M.; Hedges, S.B.; Hilton-Taylor, C.; et al. A global reptile assessment highlights shared conservation needs of tetrapods. Nature 2022, 605, 285–290. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Shao, W.; Jiang, Y.; Yan, C.; Liao, W. Assessing Reptile Conservation Status under Global Climate Change. Biology 2024, 13, 436. [Google Scholar] [CrossRef] [PubMed]
- Ding, Z.H.; Wang, X.F.; Hao, X.; Zhang, Q.; Sun, B.J.; Du, W.G. Climate warming has divergent physiological impacts on sympatric lizards. Sci. Total Environ. 2024, 912, 168992. [Google Scholar] [CrossRef]
- Wei, J.; Zeng, Z.; Zhang, X.; Shuai, L.; Teng, L.; Yan, W.; Liu, Z. Effects of habitat desertification on the community composition of lizards. Acta Ecol. Sin. 2019, 39, 1680. [Google Scholar]
- Sun, B.; Williams, C.M.; Li, T.; Speakman, J.R.; Jin, Z.; Lu, H.; Luo, L.; Du, W. Higher Metabolic Plasticity in Temperate Compared to Tropical Lizards Suggests Increased Resilience to Climate Change. Ecol. Monogr. 2022, 92, e1512. [Google Scholar] [CrossRef]
- Wang, Z.; Chen, S.; Zheng, Y.; Wu, R.; Yang, Y. Digestive tract morphology and argyrophil cell distribution of three sympatric lizard species. Eur. Zool. J. 2024, 91, 649–661. [Google Scholar] [CrossRef]
- Chen, D.L.; Li, J.; Guo, X.G. Next-generation sequencing yields a nearly complete mitochondrial genome of The Yarkand toad-headed agama (Phrynocephalus axillaris) from the Turpan Depression. Mitochondrial DNA B 2019, 4, 1198–1199. [Google Scholar] [CrossRef]
- Liang, Q.; Shi, L. Species divergence in valleys: The phylogeny of Phrynocephalus forsythii complex and description of a new species. PeerJ 2024, 12, e17175. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Ding, L.; Zhao, Y.Y.; Niu, C.K.; Wang, X.N.; Zhao, W. Toad-headed Lizard (Squamata, Agamidae) as a Potential Ring Species Inferred from Population Genetic Differentiation. Asian Herpetol. Res. 2020, 11, 312. [Google Scholar] [CrossRef]
- Zhong, Q.M.; Wang, J.L. Seasonal flexibility of kidney structure and factors regulating water and salt in Eremias multiocellata. Comp. Biochem. Physiol. Part A Mol. Integr. Physiol. 2022, 274, 111301. [Google Scholar] [CrossRef]
- Zhang, Q.; Xia, L.; He, J.; Wu, Y.; Fu, J.; Yang, Q. Comparison of phylogeographic structure and population history of two Phrynocephalus species in the Tarim Basin and adjacent areas. Mol. Phylogenet. Evol. 2010, 57, 1091–1104. [Google Scholar] [CrossRef]
- Duan, J.; Nong, J.; Jiang, L.; Chen, T.; Zhong, W.; Zhao, W.; Qi, Y.; Guo, P.; Li, Y. Population Genetic Structure of a Viviparous Sand Lizard, the Phrynocephalus forsythii in the Tarim Basin, Xinjiang of China. Diversity 2023, 15, 1182. [Google Scholar] [CrossRef]
- Orlova, V.F.; Solovyeva, E.N.; Ananjeva, N.B. Taxonomic status of var. Bedriaga 1912 (Sauria, Lacertidae): Molecular evidence from historical DNA. Zootaxa 2023, 5405, 296–300. [Google Scholar] [CrossRef]
- Zhong, Q.-M.; Zheng, Y.-H.; Wang, J.-L. Seasonal flexibility of the gut structure and physiology in Eremias multiocellata. J. Comp. Physiol. B 2023, 193, 281–291. [Google Scholar] [CrossRef]
- Liu, J.-L.; Dujsebayeva, T.N.; Chirikova, M.A.; Gong, X.; Li, D.-J.; Guo, X.-G. Does the Dzungarian racerunner (Eremias dzungarica, Orlova, Poyarkov, Chirikova, Nazarov, Munkhbaatar, Munkhbayar & Terbish, 2017) occur in China? Species delimitation and identification with DNA barcoding and morphometric analyses. Zool. Res. 2021, 42, 287–293. [Google Scholar] [CrossRef]
- Ma, L.J.; Lin, C.; Zhu, L.J.; Mahmut, P.T.G.L.; Pu, H.W. Comparison of the amino acid and extract about different subspecies of Eremias multiocellata from Xinjiang region. J. Xinjiang Med. Univ. 2011, 34, 375–378. (In Chinese) [Google Scholar]
- Ma, L.J.; Mahmut, P.T.G.L. Determination and comparison of 14 elements in body and tail of different subspecies of Eremias multiocellata from Xinjiang. Food Sci. 2006, 27, 645–648. (In Chinese) [Google Scholar]
- Ma, L.J.; Mahmut, P.T.G.L. Comparison of total phosphatide contents in different subspecies and different parts of Eremias multiocellata from Xinjiang. Food Sci. 2008, 29, 95–97. (In Chinese) [Google Scholar]
- Shi, L.; Yang, J.; Hou, M.Z.; Zhao, H.; Dong, B.J.; Xiong, J.L.; Wang, X.J.; Wang, X.H.; Zhang, X.W.; Wang, X.L.; et al. Herpe to logical surveys of Xinjiang Uygur Autonomous Region. Sichuan J. Zool. 2007, 26, 812–818+981–984. (In Chinese) [Google Scholar]
- Excoffier, L.; Lischer, H.E. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Hohna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef]
- Lanfear, R.; Frandsen, P.B.; Wright, A.M.; Senfeld, T.; Calcott, B. PartitionFinder 2: New Methods for Selecting Partitioned Models of Evolution for Molecular and Morphological Phylogenetic Analyses. Mol. Biol. Evol. 2017, 34, 772–773. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Bouckaert, R.; Heled, J.; Kuhnert, D.; Vaughan, T.; Wu, C.H.; Xie, D.; Suchard, M.A.; Rambaut, A.; Drummond, A.J. BEAST 2: A software platform for Bayesian evolutionary analysis. PLoS Comput. Biol. 2014, 10, e1003537. [Google Scholar] [CrossRef]
- Hedges, S.B.; Marin, J.; Suleski, M.; Paymer, M.; Kumar, S. Tree of life reveals clock-like speciation and diversification. Mol. Biol. Evol. 2015, 32, 835–845. [Google Scholar] [CrossRef]
- Rooney, A.P.; Honeycutt, R.L.; Derr, J.N. Historical population size change of bowhead whales inferred from DNA sequence polymorphism data. Evolution 2001, 55, 1678–1685. [Google Scholar] [PubMed]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef]
- Mayfield, J.E.; McKenna, J.F. A-T rich sequences in vertebrate DNA. A possible explanation of q-banding in metaphase chromosomes. Chromosoma 1978, 67, 157–163. [Google Scholar] [CrossRef]
- Hochachka, P.W.; Mommsen, T.P. (Eds.) Biochemistry and Molecular Biology of Fishes; Elsevier Press: Amsterdam, The Netherlands, 1993; pp. 1–38. [Google Scholar]
- Zhao, W.-H.; Yi, S.-K.; Su, J.-X.; Zhou, Q.; Shen, J.-Z.; Li, D.-P.; Zhao, X.-Y. Genetic diversity of Triplophysa yarkandensis populations in Tarim River basin in Xinjiang. Acta Hydrobiol. Sin. 2022, 46, 364–374. [Google Scholar]
- Xiao, C.; Yang, S.-y.; Cong, P. Genetic polymorphism of mitochondrial control region in Chiloscyllium plagiosum from coastal waters of southern China. Haiyang Xuebao 2008, 30, 115–121. [Google Scholar]
- Qun, Z. Origins, Population Genetics and Comparative Phyiogeography of Two Lacertid Lizards (Eremias argus and E. brenchleyi) from China. Master’s Thesis, Nanjing Normal University, Nanjing, China, 2011. [Google Scholar]
- Yue, Q. Molecular Phylogeography of the Phrynocephalus forsythia. Master’s Thesis, Lanzhou University, Lanzhou, China, 2019. [Google Scholar]
- Orlova, V.F.; Poyarkov, N.A.; Chirikova, M.A.; Nazarov, R.A.; Munkhbaatar, M.; Munkhbayar, K.; Terbish, K. MtDNA differentiation and taxonomy of Central Asian racerunners of Eremias multiocellata-E. przewalskii species complex (Squamata, Lacertidae). Zootaxa 2017, 4282, 1–42. [Google Scholar] [CrossRef]
- Zhao, E.M.; Zhao, K.T.; Zhou, K.Y. Fauna Sinica Reptile Vol. 2 Squamata Lacertilia; Science Press: Beijing, China, 1999; pp. 220–243. [Google Scholar]
- Eremchenko, V.K.; Panfilov, A.M. Taxonomic situation of multiocellated racerunner of the “multiocellata”-complex of Kyrghyzstan and neighbour China (Sauria: Lacertidae: Eremias). Sci. New Technol. 1999, 4, 112–124, (In Russian with English Abstract). [Google Scholar]
- Dai, X.; Chen, B.; Zhang, Z.W.; Wang, Y.Z. The morpholgical reseaerch of eight species of eremias distributed in China (Lacertidae eremias). Zool. Syst. 2006, 31, 697–708. [Google Scholar]
- Cai, B.; Wang, Y.; Chen, Y.; Li, J. A revised taxonomy for Chinese reptiles. Biodivers. Sci. 2015, 23, 365–382. [Google Scholar] [CrossRef]
- Wang, S.; Liu, J.L.; Chirikova, M.A.; Zhang, B.; Guo, X.G. The complete mitochondrial genome of (Reptilia, Squamata, Lacertidae) from Kyrgyzstan. Mitochondrial DNA B 2022, 7, 443–445. [Google Scholar] [CrossRef]
- Feng, S.H.; Bai, M.; Rivas-González, I.; Li, C.; Liu, S.P.; Tong, Y.J.; Yang, H.D.; Chen, G.J.; Xie, D.; Sears, K.E.; et al. Incomplete lineage sorting and phenotypic evolution in marsupials. Cell 2022, 185, 1646–1660.e18. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.-R.; Liu, X.-Y.; He, X.-T.; Wang, Y.-Y. Analysis of factors affecting animal phylogeographic patterns in China. Chin. Bull. Life Sci. 2019, 31, 968–974. [Google Scholar]
- Yan, S.; Mu, G. The Environmental Evolution of The Tarim Basin in Late Cenozoic Era. Arid. Land Geogr. 1990, 13, 1–9. [Google Scholar]
- Zhang, B.; Li, L.; Liu, X.; He, N.; Du, B.; Song, Y.; Chang, H. Link between development of the northeast Pamir Plateau and climate changes in the Tarim Basin. J. Earth Environ. 2016, 7, 346–356. [Google Scholar]
- Zhang, Q.; Tao, J.; Huang, C.; Gu, C. Discovery of Late Cenozoic Plant Fossils from the Central Kunlun Mountains. Chin. Sci. Bull. 1990, 1, 51–53. (In Chinese) [Google Scholar]
- Ren, J. Intergating Phylogegraphy and Landscape Genetics Reveals Formation Mechanisms of Genetic Variation and Its Spatial Distribution Pattern for Stipa Breviflora. Master’s Thesis, Mongolia University (Inner Mongolia Autonomous Region), Ulan Bator, Mongolia, 2019. [Google Scholar]
- Fu, B.; Awata, Y. Displacement and timing of left-lateral faulting in the Kunlun Fault Zone, northern Tibet, inferred from geologic and geomorphic features. J. Asian Earth Sci. 2007, 29, 253–265. [Google Scholar] [CrossRef]
- Burtman, V.S. Cenozoic crustal shortening between the Pamir and Tien Shanand a reconstruction of the Pamir–Tien Shan transition zonefor the Cretaceous and Palaeogene. Tectonophysics 2000, 319, 69–92. [Google Scholar] [CrossRef]
- Wan, L. Molecular Phylogeography of Chinese Eremias. Master’s Thesis, Lanzhou University, Lanzhou, China, 2006. [Google Scholar]
- Yang, S.-J.; Yin, Z.-H.; Ma, X.-M.; Lei, F.-M. Phylogeography of ground tit (Pseudopodoces humilis) based on mtDNA: Evidence of past fragmentation on the Tibetan Plateau. Mol. Phylogenet. Evol. 2006, 41, 257–265. [Google Scholar] [CrossRef]
- Wang, S.; Gao, C. The formation and evolution of the arid environment of theinland Tarim Inland Basin Cenozoic ERA. Quat. Sci. 1990, 10, 372–380. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sampling Sites | N | Combined mtDNA | ND2 | Cytb | ATP6 | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| h ± s.d. (Nh) | π ± s.d. | h ± s.d. (Nh) | π ± s.d. | h ± s.d. (Nh) | π ± s.d. | h ± s.d. (Nh) | π ± s.d. | |||
| Wuqia | WQ | 26 | 0.994 ± 0.013 (24) | 0.011 ± 0.006 | 0.775 ± 0.078 (10) | 0.024 ± 0.012 | 0.754 ± 0.072 (7) | 0.002 ± 0.001 | 0.932 ± 0.028 (14) | 0.007 ± 0.004 |
| Minfeng | MF | 19 | 0.947 ± 0.033 (13) | 0.008 ± 0.004 | 0.731 ± 0.086 (7) | 0.007 ± 0.004 | 0.819 ± 0.069 (9) | 0.010 ± 0.005 | 0.731 ± 0.066 (5) | 0.009 ± 0.005 |
| Yutian | YT | 1 | 1.000 ± 0.000 (1) | 0.000 ± 0.000 | 1.000 ± 0.000 (1) | 0.000 ± 0.000 | 1.000 ± 0.000 (1) | 0.000 ± 0.000 | 1.000 ± 0.000 (1) | 0.000 ± 0.000 |
| Hetian | HT | 12 | 0.955 ± 0.057 (10) | 0.014 ± 0.008 | 0.894 ± 0.078 (8) | 0.023 ± 0.012 | 0.894 ± 0.078 (8) | 0.013 ± 0.007 | 0.530 ± 0.136 (3) | 0.006 ± 0.004 |
| Yecheng | YC | 17 | 0.978 ± 0.027 (14) | 0.015 ± 0.008 | 0.956 ± 0.033 (12) | 0.029 ± 0.015 | 0.956 ± 0.033 (12) | 0.007 ± 0.004 | 0.853 ± 0.053 (7) | 0.006 ± 0.003 |
| Yinjisha | YJS | 10 | 0.933 ± 0.077 (8) | 0.013 ± 0.007 | 0.800 ± 0.100 (5) | 0.027 ± 0.015 | 0.533 ± 0.180 (4) | 0.003 ± 0.002 | 0.533 ± 0.180 (4) | 0.008 ± 0.005 |
| Kashi | KS | 14 | 0.956 ± 0.045 (11) | 0.017 ± 0.009 | 0.868 ± 0.068 (8) | 0.029 ± 0.015 | 0.824 ± 0.098 (8) | 0.010 ± 0.006 | 0.890 ± 0.081 (10) | 0.010 ± 0.006 |
| Tashiku-ergan | TSKEG | 14 | 0.989 ± 0.031 (13) | 0.011 ± 0.006 | 0.934 ± 0.051 (10) | 0.026 ± 0.014 | 0.506 ± 0.158 (5) | 0.003 ± 0.002 | 0.396 ± 0.159 (4) | 0.001 ± 0.001 |
| Bachu | BC | 2 | 1.000 ± 0.500 (2) | 0.012 ± 0.012 | 1.000 ± 0.500 (2) | 0.012 ± 0.012 | 1.000 ± 0.500 (2) | 0.010 ± 0.011 | 1.000 ± 0.500 (2) | 0.014 ± 0.015 |
| Total | 115 | 0.996 ± 0.002 (96) | 0.019 ± 0.009 | 0.974 ± 0.006 (61) | 0.029 ± 0.014 | 0.965 ± 0.007 (50) | 0.014 ± 0.007 | 0.961 ± 0.006 (39) | 0.013 ± 0.007 | |
| Primers | Primer Sequence | Tm |
|---|---|---|
| 1F-ATP6 | 5′-CCGCTATCTCTAAGCATGAAAGT-3′ | 58 °C |
| 1R-ATP6 | 5′-TGTTACCCTGGGAGTTCCAC-3′ | |
| 48F-ND2A | 5′-TGTAATCATCACCGCCACCA-3′ | 58 °C |
| 557R-ND2A | 5′-TGTGCGATTGATGAGTAGGCT-3′ | |
| 28F-ND2B | 5′-TCGCCCTCACCTCTACCTTA-3′ | 58 °C |
| 537R-ND2B | 5′-CGGTTGTGTTGAGGGGTTTT-3′ | |
| 11F-Cytb | 5′-ACATACGAAAACAACATCCAATCT-3′ | 58 °C |
| 1127R-Cytb | 5′-TTATTTTCTAGGGTGGCGGCT-3′ |
| Pairwise | Divergence Time | Median Time |
|---|---|---|
| E. multiocellata and D. daghestanica | 36.1–79.0 MYA | 66 MYA |
| E. multiocellata and L. agilis | 36.1–79.0 MYA | 66 MYA |
| E. multiocellata and E. brenchleyi | 28.5–47.3 MYA | 34 MYA |
| E. multiocellata and E. argus | 28.5–47.3 MYA | 34 MYA |
| B1 | B2 | B3 | M1 | M2 | M3 | |
|---|---|---|---|---|---|---|
| B1 | - | - | - | - | - | - |
| B2 | 0.0266 | - | - | - | - | |
| B3 | 0.0144 | 0.0278 | - | - | - | - |
| M1 | - | - | - | - | - | - |
| M2 | - | - | 0.0268 | - | - | |
| M3 | - | - | - | 0.0130 | 0.0280 | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, J.; Yan, H.; Chen, T.; Chen, W.; Chen, Y.; Zhong, W.; Qi, Y.; Zhao, W.; Li, Y. Phylogeographic Analyses of the Viviparous Multiocellated Racerunner (Eremias multiocellata) in the Tarim Basin of China. Diversity 2025, 17, 313. https://doi.org/10.3390/d17050313
Zhang J, Yan H, Chen T, Chen W, Chen Y, Zhong W, Qi Y, Zhao W, Li Y. Phylogeographic Analyses of the Viviparous Multiocellated Racerunner (Eremias multiocellata) in the Tarim Basin of China. Diversity. 2025; 17(5):313. https://doi.org/10.3390/d17050313
Chicago/Turabian StyleZhang, Junzhe, Haifan Yan, Tianying Chen, Wenhan Chen, Yulu Chen, Wen Zhong, Yue Qi, Wei Zhao, and You Li. 2025. "Phylogeographic Analyses of the Viviparous Multiocellated Racerunner (Eremias multiocellata) in the Tarim Basin of China" Diversity 17, no. 5: 313. https://doi.org/10.3390/d17050313
APA StyleZhang, J., Yan, H., Chen, T., Chen, W., Chen, Y., Zhong, W., Qi, Y., Zhao, W., & Li, Y. (2025). Phylogeographic Analyses of the Viviparous Multiocellated Racerunner (Eremias multiocellata) in the Tarim Basin of China. Diversity, 17(5), 313. https://doi.org/10.3390/d17050313