
Two Complete Mitochondrial Genomes of Potamanthidae (Ephemeroptera): Genome Features and Phylogenetic Implications


Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection and Species Identification
2.2. DNA Extraction and Mitogenome Sequencing
2.3. Mitogenome Assembly and Bioinformatic Analysis
2.4. Mitogenome Assembly and Bioinformatic Analysis
3. Results and Discussion
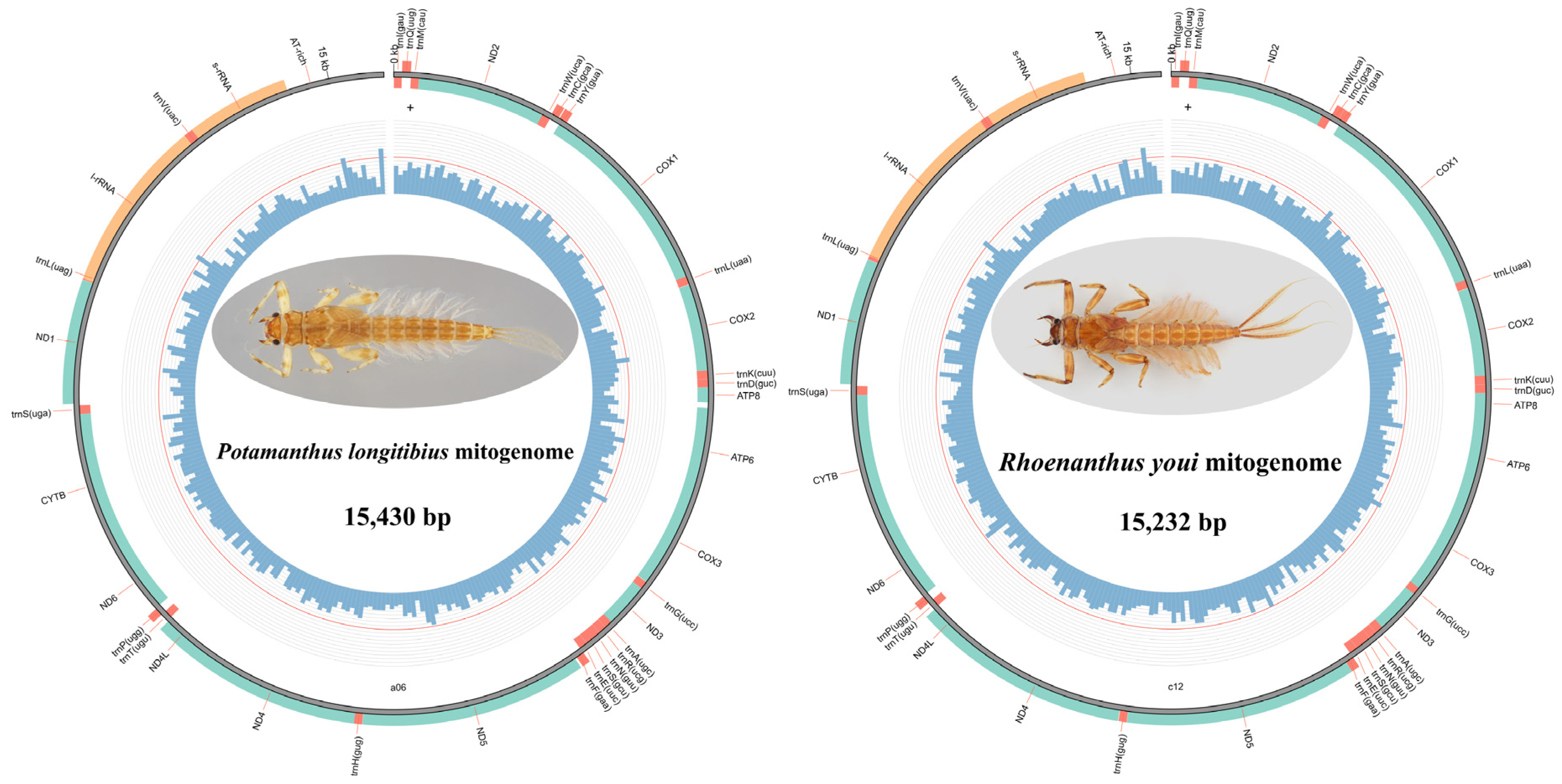
3.1. Genome Organization
3.2. Nucleotide Composition
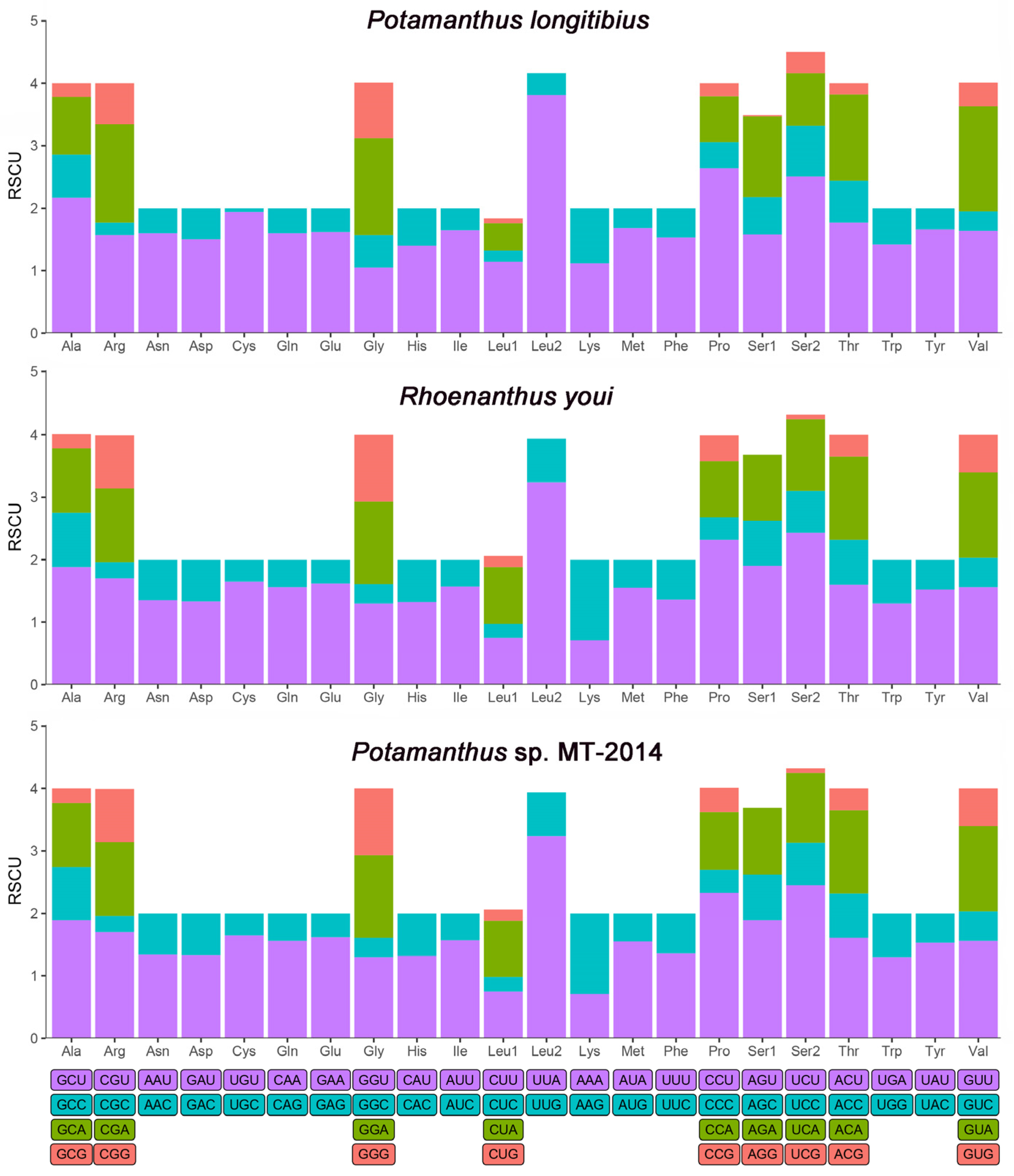
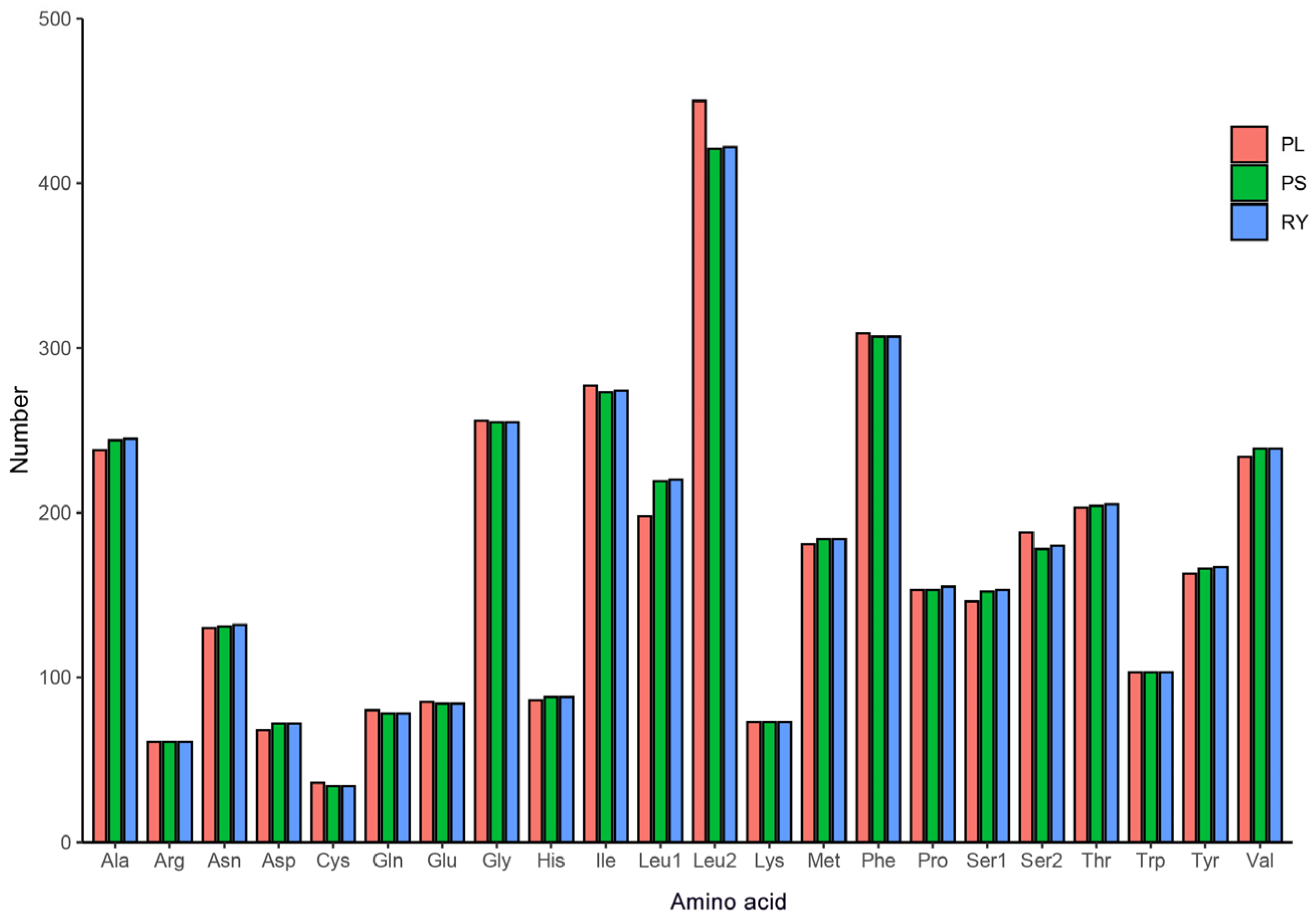
3.3. Protein-Coding Genes and Codon Usage
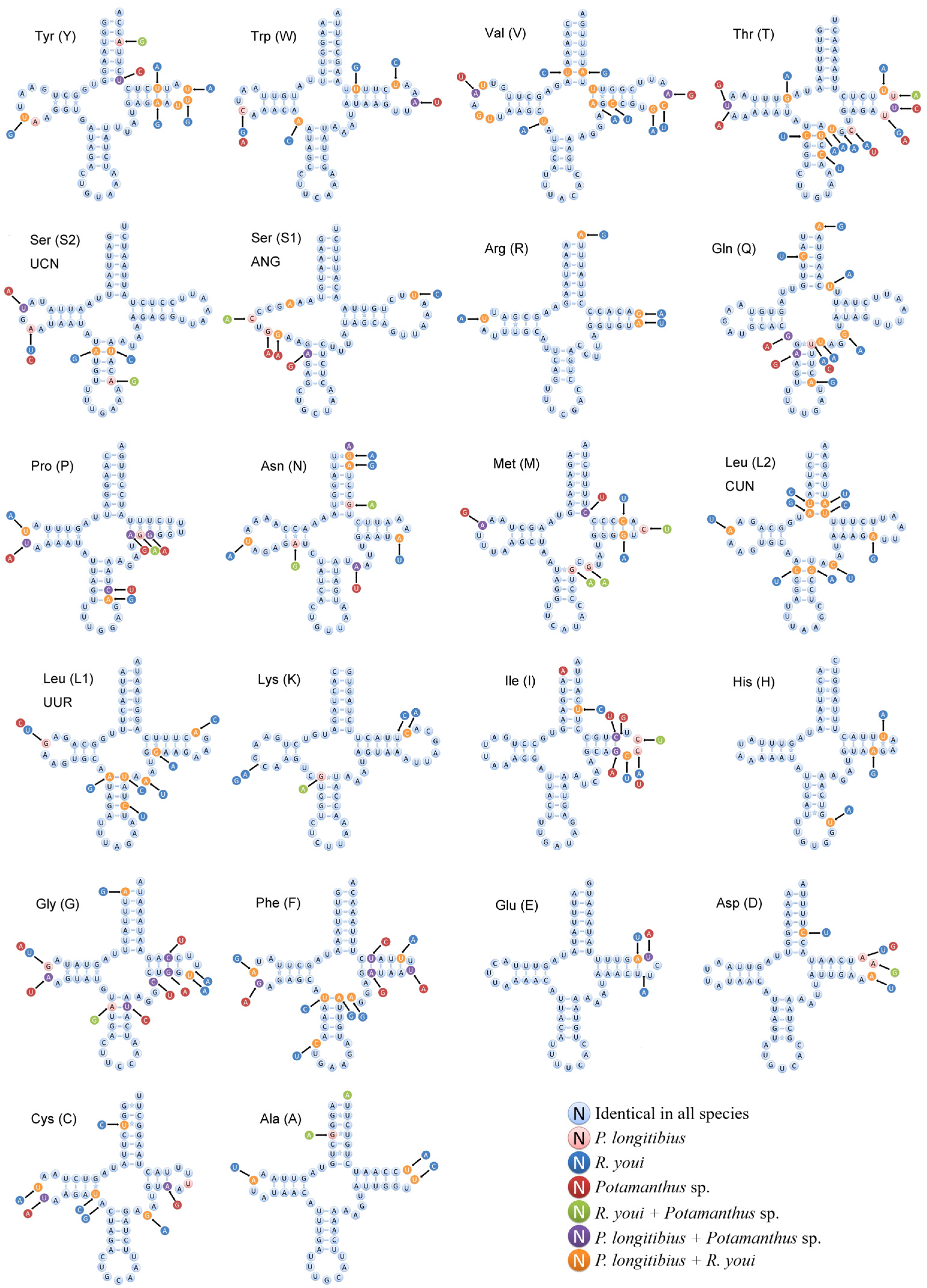
3.4. Ribosomal and Transfer RNA Genes
3.5. Non-Coding Regions
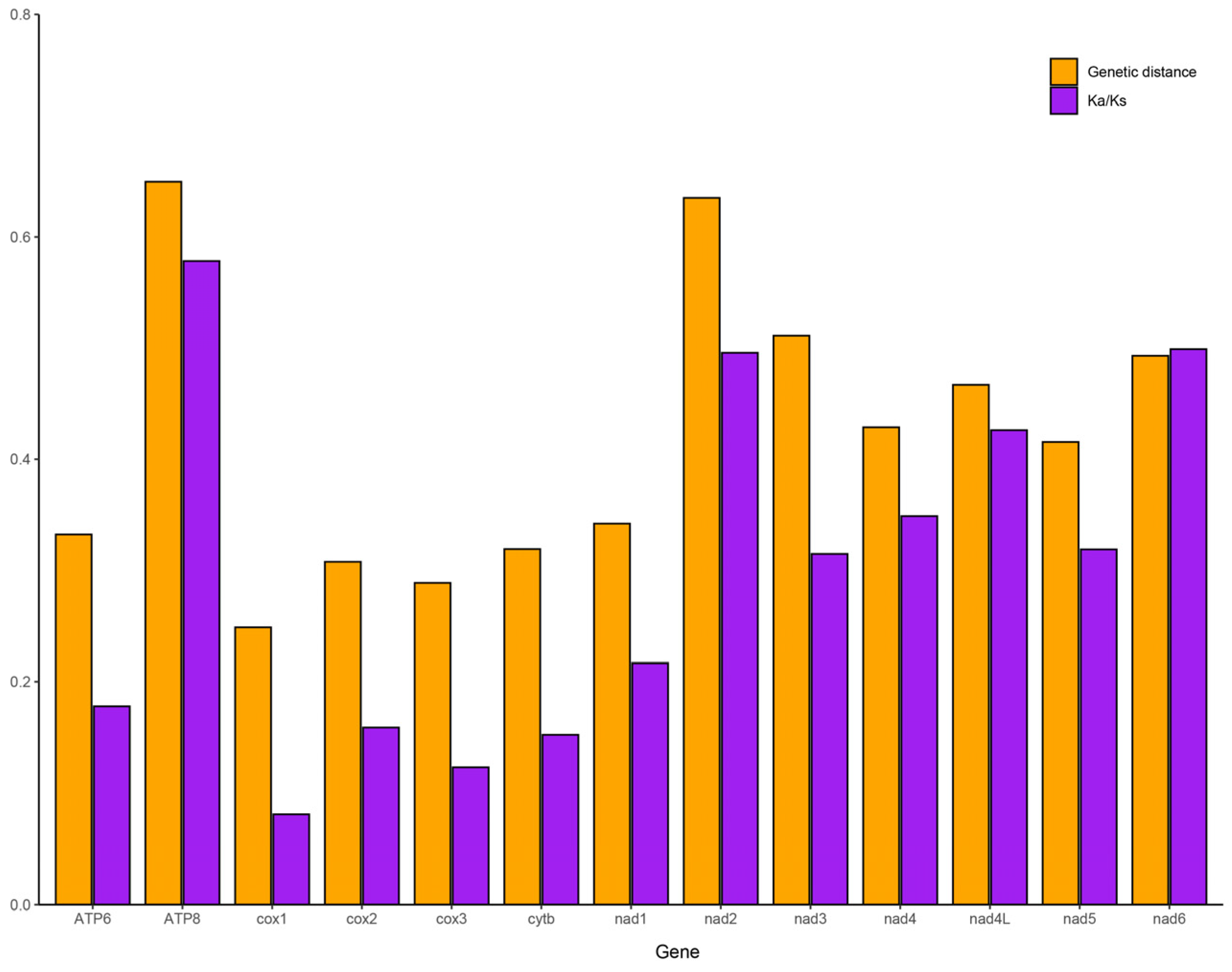
3.6. Phylogenetic Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bernt, M.; Braband, A.; Schierwater, B.; Stadler, P.F. Genetic aspects of mitochondrial genome evolution. Mol. Phylogenet. Evol. 2013, 69, 328–338. [Google Scholar] [CrossRef] [PubMed]
- Boore, J.L. Animal mitochondrial genomes. Nucleic Acids Res. 1999, 27, 1767–1780. [Google Scholar] [CrossRef] [PubMed]
- Burger, G.; Gray, M.W.; Lang, B.F. Mitochondrial genomes: Anything goes. Trends Genet. 2003, 19, 709–716. [Google Scholar] [CrossRef] [PubMed]
- Wolstenholme, D.R. Animal mitochondrial DNA: Structure and evolution. Int. Rev. Cytol. 1992, 141, 173–216. [Google Scholar]
- Simon, S.; Hadrys, H. A comparative analysis of complete mitochondrial genomes among Hexapoda. Mol. Phylogenet. Evol. 2013, 69, 393–403. [Google Scholar] [CrossRef]
- Dong, X.; Yi, W.; Zheng, C.; Zhu, X.; Wang, S.; Xue, H.; Ye, Z.; Bu, W. Species delimitation of rice seed bugs complex: Insights from mitochondrial genomes and ddRAD-seq data. Zool. Scr. 2022, 51, 185–198. [Google Scholar] [CrossRef]
- Ge, X.; Wang, J.; Zang, H.; Chai, L.; Liu, W.; Zhang, J.; Yan, C.; Wang, B. Mitogenomics provide new phylogenetic insights of the family Apataniidae (Trichoptera: Integripalpia). Insects 2024, 15, 973. [Google Scholar] [CrossRef]
- Bunmee, K.; Thaenkham, U.; Saralamba, N.; Ponlawat, A.; Zhong, D.; Cui, L.; Sattabongkot, J.; Sriwichai, P. Population genetic structure of the malaria vector Anopheles minimus in Thailand based on mitochondrial DNA markers. Parasites Vectors 2021, 14, 496. [Google Scholar] [CrossRef]
- Chang, H.; Qiu, Z.; Yuan, H.; Wang, X.; Li, X.; Sun, H.; Guo, X.; Lu, Y.; Feng, X.; Majid, M.; et al. Evolutionary rates of and selective constraints on the mitochondrial genomes of Orthoptera insects with different wing types. Mol. Phylogenet. Evol. 2020, 145, 106734. [Google Scholar] [CrossRef]
- Ge, X.; Peng, L.; Vogler, A.P.; Morse, J.C.; Yang, L.; Sun, C.; Wang, B. Massive gene rearrangements of mitochondrial genomes and implications for the phylogeny of Trichoptera (Insecta). Syst. Entomol. 2023, 48, 278–295. [Google Scholar] [CrossRef]
- Cameron, S.L. Insect mitochondrial genomics: A decade of progress. Annu. Rev. Entomol. 2024, 70, 83–101. [Google Scholar] [CrossRef] [PubMed]
- Jacobus, L.M.; Macadam, C.R.; Sartori, M. Mayflies (Ephemeroptera) and their contributions to ecosystem services. Insects 2019, 10, 170. [Google Scholar] [CrossRef] [PubMed]
- Jacobus, L.M.; Salles, F.F.; Price, B.; Pereira-Da-Conceicoa, L.; Dominguez, E.; Suter, P.J.; Molineri, C.; Tiunova, T.M.; Sartori, M. Mayfly taxonomy (Arthropoda: Hexapoda: Ephemeroptera) during the first two decades of the twenty-first century and the concentration of taxonomic publishing. Zootaxa 2021, 4979, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Sartori, M.; Brittain, J.E. Order Ephemeroptera. In Ecology and General Biology, Vol I: Thorp and Covich’s Freshwater Invertebrates, 4th ed.; Thorp, J.H., Rogers, D.C., Eds.; Academic Press: New York, NY, USA, 2015; pp. 873–891. [Google Scholar]
- Lei, Z.; Li, M.; Deng, M.; Zhou, C. First description of the Chinese mayfly Ephemera lota Navás, 1934 (Ephemeroptera: Ephemeridae) based on imagines with designation of the species neotype. Aquat. Insects 2024, 45, 319–334. [Google Scholar] [CrossRef]
- Ogden, T.H.; Gattolliat, J.L.; Sartori, M.; Staniczek, A.H.; Soldán, T.; Whiting, M.F. Towards a new paradigm in mayfly phylogeny (Ephemeroptera): Combined analysis of morphological and molecular data. Syst. Entomol. 2009, 34, 616–634. [Google Scholar] [CrossRef]
- Miller, D.B.; Bartlett, S.; Sartori, M.; Breinholt, J.W.; Ogden, T.H. Anchored phylogenomics of burrowing mayflies (Ephemeroptera) and the evolution of tusks. Syst. Entomol. 2018, 43, 692–701. [Google Scholar] [CrossRef]
- Ogden, T.H.; Breinholt, J.W.; Bybee, S.M.; Miller, D.B.; Sartori, M.; Shiozawa, D.; Whiting, M.F. Mayfly phylogenomics: Initial evaluation of anchored hybrid enrichment data for the order Ephemeroptera. Zoosymposia 2019, 16, 167–181. [Google Scholar]
- Guo, Z.Q.; Shen, C.Y.; Cheng, H.Y.; Chen, Y.X.; Wu, H.Y.; Storey, K.B.; Yu, D.N.; Zhang, J.Y. Mitogenome-based phylogeny with divergence time estimates revealed the presence of cryptic species within Heptageniidae (Insecta, Ephemeroptera). Insects 2024, 15, 745. [Google Scholar] [CrossRef]
- Li, R.; Zhang, W.; Ma, Z.X.; Zhou, C.F. Novel gene rearrangement pattern in the mitochondrial genomes of Torleya mikhaili and Cincticostella fusca (Ephemeroptera: Ephemerellidae). Int. J. Biol. Macromol. 2020, 165, 3106–3114. [Google Scholar] [CrossRef]
- Bae, Y.J.; McCafferty, W.P. Phylogenetic systematics of the Potamanthidae (Ephemeroptera). Trans. Am. Entomol. Soc. 1991, 117, 1–143. [Google Scholar]
- Kwanboon, S.; Sartori, M.; Boonsoong, B. Behningiidae and Potamanthidae (Insecta, Ephemeroptera) in Thailand. ZooKeys 2021, 1067, 57. [Google Scholar] [CrossRef] [PubMed]
- Bartholomae, P.G.; Meier, P.G. Notes on the life history of Potamanthus myops in southeastern Michigan (Ephemeroptera: Potamanthidae). Great Lakes Entomol. 1977, 10, 10. [Google Scholar] [CrossRef]
- Han, N.A.; Zhang, M.; Zhou, C.F. The genus Rhoenanthus Eaton, 1881 in China with the redescription of R. hunanensis You & Gui, 1995 (Ephemeroptera: Potamanthidae). Zootaxa 2021, 4903, zootaxa-4903. [Google Scholar]
- Li, W.J.; Zhou, C.F. A detailed comparison of two species in the genus Potamanthus Pictet, 1843 from China (Ephemeroptera, Potamanthidae). ZooKeys 2022, 1125, 193–205. [Google Scholar] [CrossRef]
- Li, W.J.; Mu, P.X.; Zhou, C.F. Systematics of Potamanthodes nanchangi Hsu, 1936 (Ephemeroptera: Potamanthidae). Zootaxa 2022, 5213, 149–158. [Google Scholar] [CrossRef]
- Wingett, S.W.; Andrews, S. FastQ Screen: A tool for multi-genome mapping and quality control. F1000Research 2018, 7, 1338. [Google Scholar] [CrossRef]
- Patel, R.K.; Jain, M. NGS QC Toolkit: A toolkit for quality control of next-generation sequencing data. PLoS ONE 2012, 7, e30619. [Google Scholar] [CrossRef]
- Schmieder, R.; Edwards, R. Quality control and preprocessing of metagenomic datasets. Bioinformatics 2011, 27, 863–864. [Google Scholar] [CrossRef]
- Nicolas, D.; Patrick, M.; Guillaume, S. NOVOPlasty: De novo assembly of organelle genomes from whole-genome data. Nucleic Acids Res. 2017, 45, e18. [Google Scholar]
- Meng, G.; Li, Y.; Yang, C.; Liu, S. MitoZ: A toolkit for mitochondrial genome assembly, annotation, and visualization. Nucleic Acids Res. 2019, 47, e63. [Google Scholar] [CrossRef]
- Laslett, D.; Canbäck, B. ARWEN, a program to detect tRNA genes in metazoan mitochondrial nucleotide sequences. Bioinformatics 2008, 24, 172–175. [Google Scholar] [CrossRef] [PubMed]
- Lowe, T.M.; Chan, P.P. tRNAscan-SE On-line: Integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 2016, 44, W54–W57. [Google Scholar] [CrossRef] [PubMed]
- Bernt, M.; Donath, A.; Jühling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Pütz, J.; Middendorf, M.; Stadler, P.F. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Frazer, K.A.; Pachter, L.; Poliakov, A.; Rubin, E.M.; Dubchak, I. VISTA: Computational tools for comparative genomics. Nucleic Acids Res. 2004, 32, W273–W279. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Perna, N.T.; Kocher, T.D. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J. Mol. Evol. 1995, 41, 353–358. [Google Scholar] [CrossRef]
- Zhang, D.; Gao, F.; Jakovlić, I.; Zhou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef]
- Katoh, K.; Kuma, K.; Toh, H.; Miyata, T. MAFFT version 5: Improvement in accuracy of multiple sequence alignment. Nucleic Acids Res. 2005, 33, 511–518. [Google Scholar] [CrossRef]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. TrimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef]
- Kück, P.; Longo, G.C. FASconCAT-G: Extensive functions for multiple sequence alignment preparations concerning phylogenetic studies. Front. Zool. 2014, 11, 81. [Google Scholar] [CrossRef] [PubMed]
- Lanfear, R.; Calcott, B.; Ho, S.Y.; Guindon, S. PartitionFinder: Combined selection of partitioning schemes and substitution models for phylogenetic analyses. Mol. Biol. Evol. 2012, 29, 1695–1701. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.; Pfeiffer, W.T.; Schwartz, T. Creating the CIPRES science gateway for inference of large phylogenetic Trees. In Proceedings of the Gateway Computing Environments Workshop (GCE), New Orleans, LA, USA, 14 November 2010; Institute of Electrical and Electronics Engineers (IEEE): New Orleans, LA, USA, 2010; pp. 1–8. [Google Scholar]
- Rambaut, A. FigTree v1. 4.2: Tree Figure Drawing Tool. Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 12 October 2024).
- Clary, D.O.; Wolstenholme, D.R. The mitochondrial DNA molecule of Drosophila yakuba: Nucleotide sequence, gene organization, and genetic code. J. Mol. Evol. 1985, 22, 252–271. [Google Scholar] [CrossRef]
- Dai, J.; Liu, M.; Di Giulio, A.; Sabatelli, S.; Wang, W.; Audisio, P. The first two complete mitochondrial genomes for the subfamily Meligethinae (Coleoptera: Nitidulidae) and implications for the higher phylogeny of Nitidulidae. Insects 2024, 15, 57. [Google Scholar] [CrossRef]
- Ojala, D.; Montoya, J.; Attardi, G. tRNA punctuation model of RNA processing in human mitochondria. Nature 1981, 290, 470–474. [Google Scholar] [CrossRef]
- Xue, C.; Zhang, D.; Liu, D.K.; Jiao, L.Z.; Li, R.; Yi, X.F. First record of the complete mitochondrial genome for the genus Borbo (Lepidoptera, Hesperiidae): Characterization and comparative genomic analysis. Diversity 2024, 16, 560. [Google Scholar] [CrossRef]
- Zhang, L.; Li, W.H. Mammalian housekeeping genes evolve more slowly than tissue-specific genes. Mol. Biol. Evol. 2004, 21, 236–239. [Google Scholar] [CrossRef]
- Cameron, S.L. Insect mitochondrial genomics: Implications for evolution and phylogeny. Annu. Rev. Entomol. 2014, 59, 95–117. [Google Scholar] [CrossRef]
- Varani, G.; McClain, W.H. The G-U wobble base pair: A fundamental building block of RNA structure crucial to RNA function in diverse biological systems. EMBO Rep. 2000, 1, 18–23. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Shu, X.H.; Li, X.D.; Meng, L.; Li, B.P. Comparative mitogenome analysis of three species and monophyletic inference of Catantopinae (Orthoptera: Acridoidea). Genomics 2019, 111, 1728–1735. [Google Scholar] [CrossRef] [PubMed]
- Lv, C.; Li, Q.; Kong, L. Comparative analyses of the complete mitochondrial genomes of Dosinia clams and their phylogenetic position within Veneridae. PLoS ONE 2018, 13, e0196466. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.D.; Jia, Y.Y.; Dai, X.Y.; Ma, J.L.; Storey, K.B.; Zhang, J.Y.; Yu, D.N. The mitochondrial genome of Caenis sp. (Ephemeroptera: Caenidae) from Fujian and the phylogeny of Caenidae within Ephemeroptera. Mitochondrial DNA Part B 2019, 5, 192–193. [Google Scholar] [CrossRef]
- Guan, J.Y.; Zhang, Z.Y.; Cao, Y.R.; Xu, X.D.; Storey, K.B.; Yu, D.N.; Zhang, J.Y. The complete mitochondrial genome of Choroterpes (Euthralus) yixingensis (Ephemeroptera: Leptophlebiidae) and its mitochondrial protein-coding gene expression under imidacloprid stress. Gene 2021, 800, 145833. [Google Scholar] [CrossRef]
- Yu, D.N.; Yu, P.P.; Zhang, L.P.; Storey, K.B.; Gao, X.Y.; Zhang, J.Y. Increasing 28 mitogenomes of Ephemeroptera, Odonata and Plecoptera support the Chiastomyaria hypothesis with three different outgroup combinations. PeerJ 2021, 9, e11402. [Google Scholar] [CrossRef]
- Li, R.; Ma, Z.X.; Zhou, C.F. The first two complete mitochondrial genomes of Neoephemeridae (Ephemeroptera): Comparative analysis and phylogenetic implication for Furcatergalia. Genes 2021, 12, 1875. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Strand | Position | Intergenic Nucleotides | Codon | Anticodon | |||
|---|---|---|---|---|---|---|---|---|
| Plon | Ryou | Plon | Ryou | Plon | Ryou | |||
| trnI | H | 1–63 | 1–63 | 0 | 3 | - | - | GAU |
| trnQ | L | 64–132 | 67–135 | 1 | 5 | - | - | UUG |
| trnM | H | 134–198 | 141–205 | 0 | 0 | - | - | CAU |
| ND2 | H | 199–1215 | 206–1222 | −2 | −2 | ATC/TAA | ATT/TAA | |
| trnW | H | 1214–1281 | 1221–1288 | −8 | −8 | - | - | UCA |
| trnC | L | 1274–1335 | 1281–1341 | 7 | 0 | - | - | GCA |
| trnY | L | 1343–1408 | 1342–1405 | 1 | 1 | - | - | GUA |
| COX1 | H | 1410–2945 | 1407–2942 | −5 | −5 | CGA/TAA | CGA/TAA | |
| trnL | H | 2941–3006 | 2938–3003 | 2 | 2 | - | - | UAA |
| COX2 | H | 3009–3696 | 3006–3693 | 0 | 0 | ATG/T | ATG/T | |
| trnK | H | 3697–3764 | 3694–3763 | 0 | 0 | - | - | CUU |
| trnD | H | 3765–3829 | 3764–3828 | 0 | 0 | - | - | GUC |
| ATP8 | H | 3830–3994 | 3829–3990 | −7 | −7 | ATT/TAA | ATC/TAA | |
| ATP6 | H | 3988–4665 | 3984–4661 | −1 | −1 | ATG/TAA | ATG/TAA | |
| COX3 | H | 4665–5453 | 4661–5464 | 2 | −17 | ATG/TAA | ATG/TAA | |
| trnG | H | 5456–5517 | 5448–5510 | 0 | 0 | - | - | UCC |
| ND3 | H | 5518–5871 | 5511–5864 | −1 | −1 | ATA/TAA | ATC/TAA | |
| trnA | H | 5871–5933 | 5864–5927 | 0 | 0 | - | - | UGC |
| trnR | H | 5934–5996 | 5928–5990 | 0 | 0 | - | - | UCG |
| trnN | H | 5997–6060 | 5991–6054 | −1 | −3 | - | - | GUU |
| trnS | H | 6060–6126 | 6052–6117 | 0 | 0 | - | - | GCU |
| trnE | H | 6127–6191 | 6118–6183 | −2 | −2 | - | - | UUC |
| trnF | L | 6190–6253 | 6182–6246 | 6 | 6 | - | - | GAA |
| ND5 | L | 6260–7988 | 6253–7981 | 1 | 1 | ATG/T | ATG/T | |
| trnH | L | 7990–8052 | 7983–8044 | −1 | 3 | - | - | GUG |
| ND4 | L | 8052–9395 | 8048–9391 | −7 | −7 | ATG/TAA | ATG/TAA | |
| ND4L | L | 9389–9685 | 9385–9681 | 7 | 2 | ATG/TAA | ATG/TAA | |
| trnT | H | 9693–9756 | 9684–9747 | 0 | 0 | - | - | UGU |
| trnP | L | 9757–9819 | 9748–9811 | 2 | 2 | - | - | UGG |
| ND6 | H | 9822–10,334 | 9814–10,329 | −1 | −1 | ATT/TAA | ATT/TAA | |
| CYTB | H | 10,334–11,468 | 10,329–11,463 | 0 | 0 | ATG/T | ATG/T | |
| trnS | H | 11,469–11,538 | 11,464–11,533 | 11 | 12 | - | - | UGA |
| ND1 | L | 11,550–12,491 | 11,546–12,487 | 1 | 1 | ATG/TAA | ATG/TAA | |
| trnL | L | 12,493–12,557 | 12,489–12,553 | 0 | 0 | - | - | UAG |
| rrnL | L | 12,558–13,818 | 12,554–13,811 | 10 | 0 | - | - | |
| trnV | L | 13,819–13,889 | 13,812–13,878 | 0 | 0 | - | - | UAC |
| rrnS | L | 13,890–14,670 | 13,879–14,652 | 0 | 0 | - | - | |
| CR | H | 14,671–15,430 | 14,653–15,232 | 0 | 0 | - | - | |
| Length | A + T % | AT-Skew | GC-Skew | |||||
|---|---|---|---|---|---|---|---|---|
| Plon | Ryou | Plon | Ryou | Plon | Ryou | Plon | Ryou | |
| Whole genome | 15,430 | 15,232 | 68.79 | 66.75 | −0.029 | −0.038 | −0.235 | −0.245 |
| PCGs | 11,187 | 11,202 | 67.91 | 65.73 | −0.201 | −0.187 | −0.004 | −0.023 |
| 1st codon | 3732 | 3737 | 60.90 | 59.93 | −0.111 | −0.083 | 0.208 | 0.178 |
| 2nd codon | 3729 | 3734 | 62.88 | 63.01 | −0.407 | −0.403 | −0.130 | −0.125 |
| 3rd codon | 3729 | 3734 | 79.99 | 74.28 | −0.107 | −0.088 | −0.185 | −0.192 |
| tRNAs | 1436 | 1434 | 70.68 | 70.57 | −0.005 | 0.026 | 0.121 | 0.137 |
| rRNAs | 2042 | 2032 | 71.65 | 69.69 | 0.023 | 0.073 | 0.323 | 0.325 |
| CR | 760 | 580 | 69.74 | 66.55 | −0.121 | −0.130 | −0.487 | −0.351 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, R.; Xue, C.; Ma, Z.; Zhou, C. Two Complete Mitochondrial Genomes of Potamanthidae (Ephemeroptera): Genome Features and Phylogenetic Implications. Diversity 2025, 17, 164. https://doi.org/10.3390/d17030164
Li R, Xue C, Ma Z, Zhou C. Two Complete Mitochondrial Genomes of Potamanthidae (Ephemeroptera): Genome Features and Phylogenetic Implications. Diversity. 2025; 17(3):164. https://doi.org/10.3390/d17030164
Chicago/Turabian StyleLi, Ran, Chao Xue, Zhenxing Ma, and Changfa Zhou. 2025. "Two Complete Mitochondrial Genomes of Potamanthidae (Ephemeroptera): Genome Features and Phylogenetic Implications" Diversity 17, no. 3: 164. https://doi.org/10.3390/d17030164
APA StyleLi, R., Xue, C., Ma, Z., & Zhou, C. (2025). Two Complete Mitochondrial Genomes of Potamanthidae (Ephemeroptera): Genome Features and Phylogenetic Implications. Diversity, 17(3), 164. https://doi.org/10.3390/d17030164