
Growth and Genome Features of Non-O1/O139 Vibrio cholerae Isolated from Three Species of Common Freshwater Fish
 , , and
, , and
Abstract
1. Introduction
2. Materials and Methods
2.1. V. cholerae Isolates and Culture Conditions
2.2. Growth of V. cholerae Isolates under Different pH and NaCI Conditions
2.3. Genome Sequencing, Assembly, and Annotation
2.4. Comparative Genome Analysis
2.5. Multilocus Sequence Typing (MLST) Analysis
2.6. Phylogenetic Tree Construction
2.7. Antibiotic and Heavy Metal Resistance Assays
3. Results and Discussion
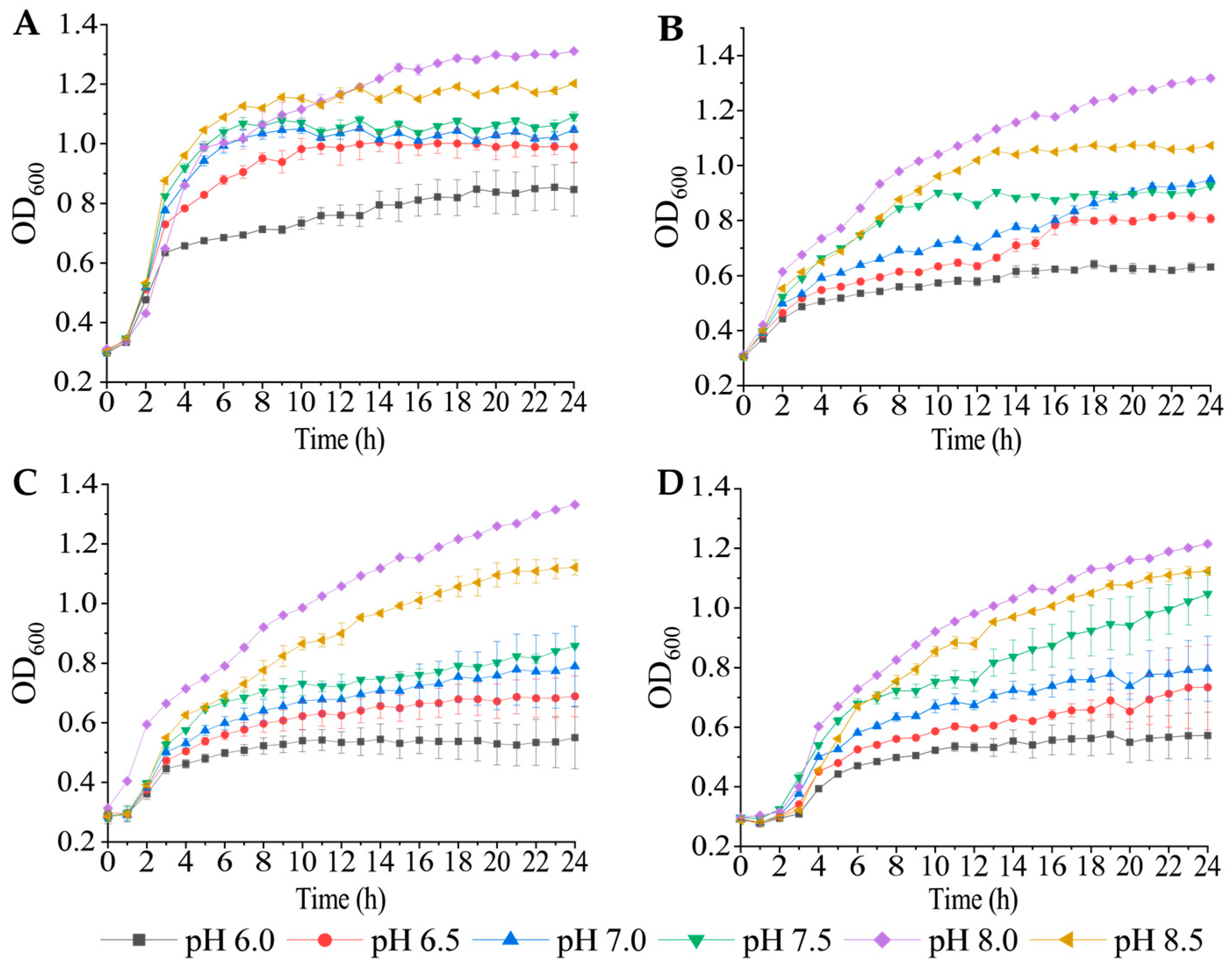
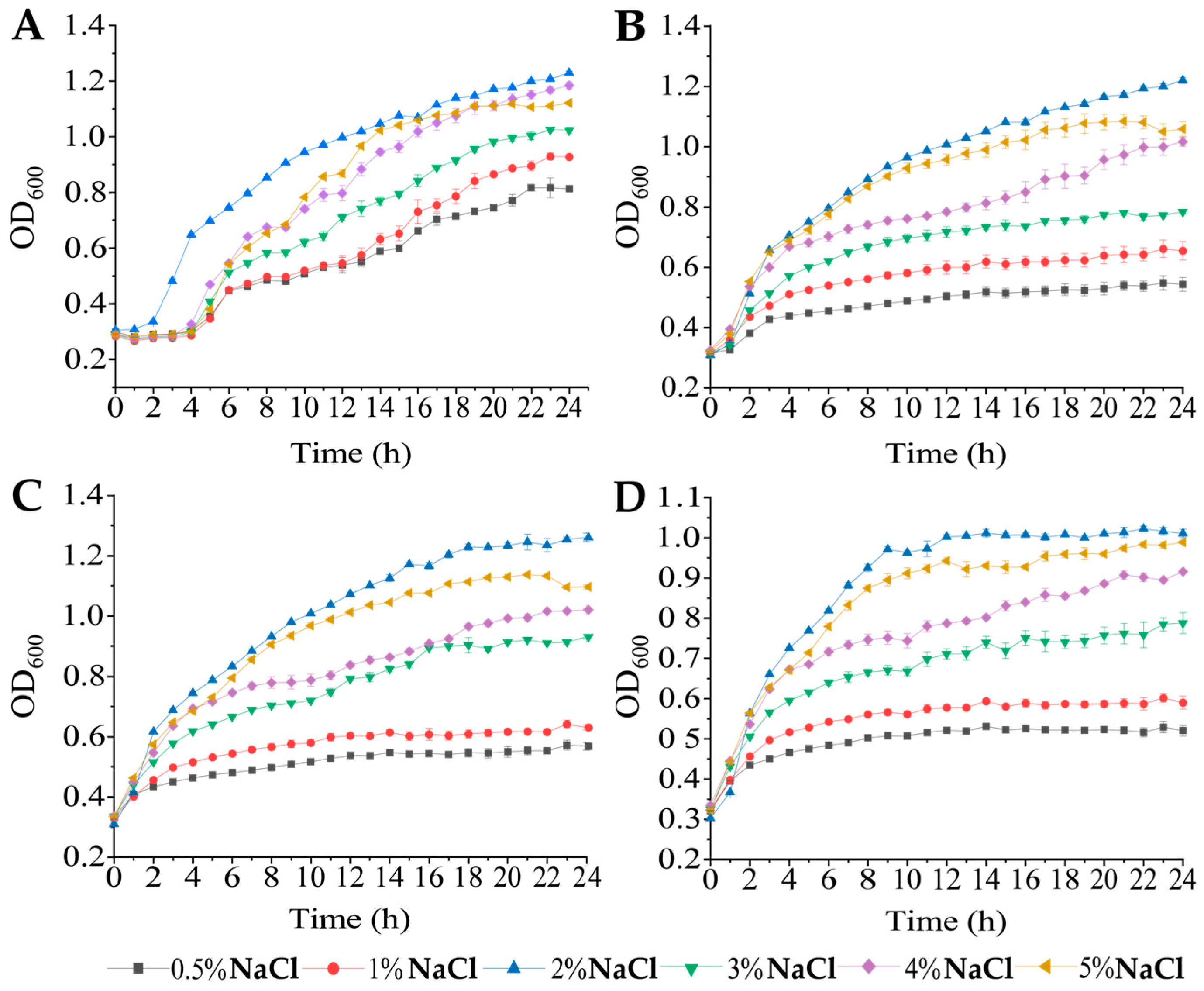
3.1. Growth Profiles of the Four V. cholerae Isolates under Different pH and NaCI Conditions
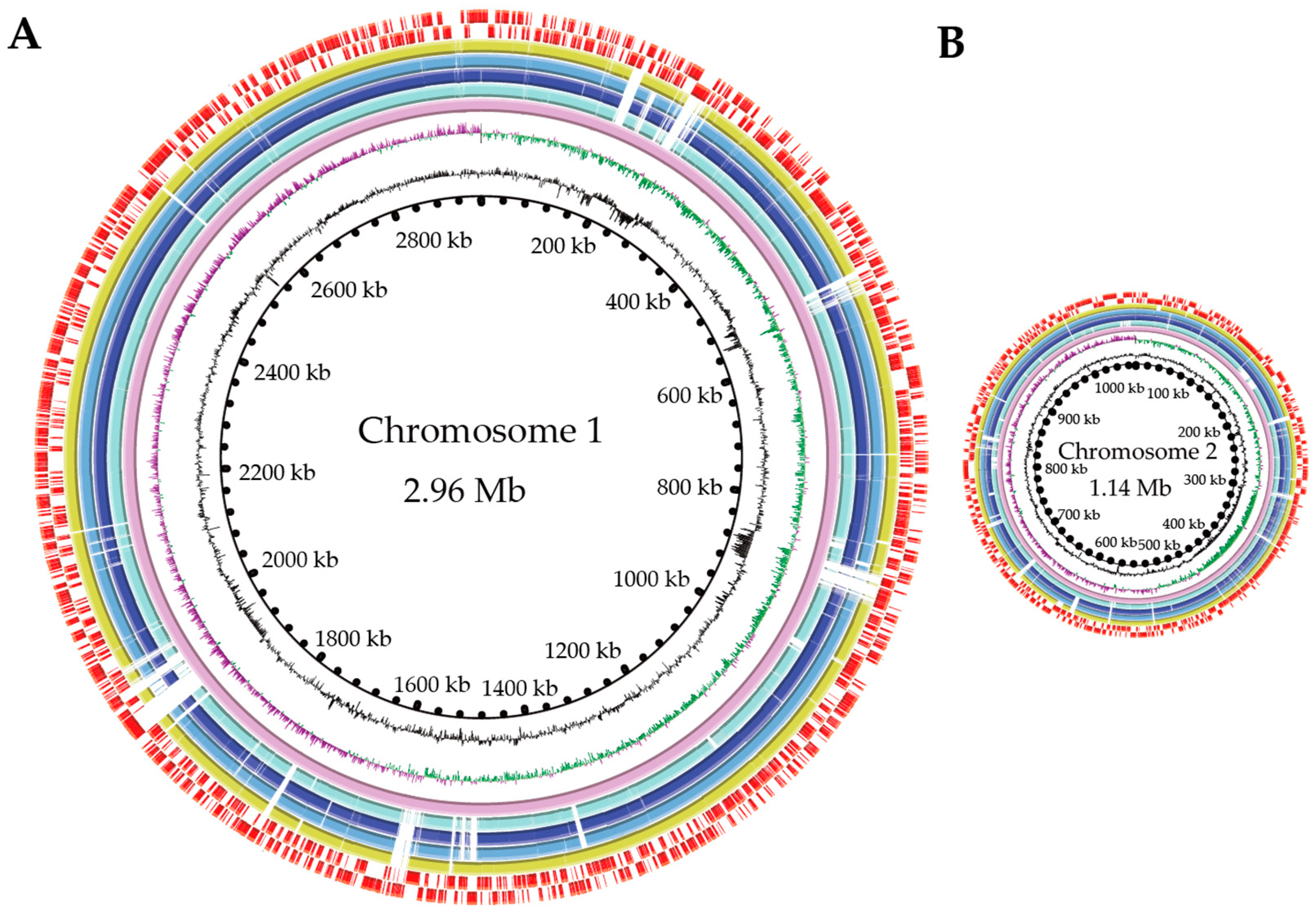
3.2. Genome Features of the Four V. cholerae Isolates from the Common Freshwater Fish
3.3. Putative MGEs in the Four V. cholerae Isolates from the Common Freshwater Fish
3.3.1. GIs
3.3.2. Prophages
3.3.3. Ins
3.3.4. ISs
3.4. CRISPR-Cas System Arrays
3.5. Putative Virulence-Associated Genes in the Four V. cholerae Genomes
3.6. Resistance-Associated Genes in the Four V. cholerae Genomes and Their Resistance Phenotypes

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibiotic Agent | Resistance-Related Gene | V. cholerae Isolate | Reference |
|---|---|---|---|
| Fluoroquinolone | crp | 7-6-5, L1-1, L10-48, B5-86 | [48] |
| QnrVC4 | 7-6-5, L1-1, B5-86 | [50] | |
| Chloramphenicol | catB9 | L10-48 | [52] |
| Carbapenem | VarG | 7-6-5, B5-86 | [51] |
| Polymyxin | AlmG, ugd | 7-6-5, L1-1, L10-48, B5-86 | [49,54] |
| beta-Lactam | CARB-7 | L10-48 | [53] |
| Nitroimidazole | msbA | 7-6-5, L1-1, L10-48, B5-86 | [55] |
| Streptogramin | vatF | 7-6-5, L1-1, L10-48, B5-86 | [56] |
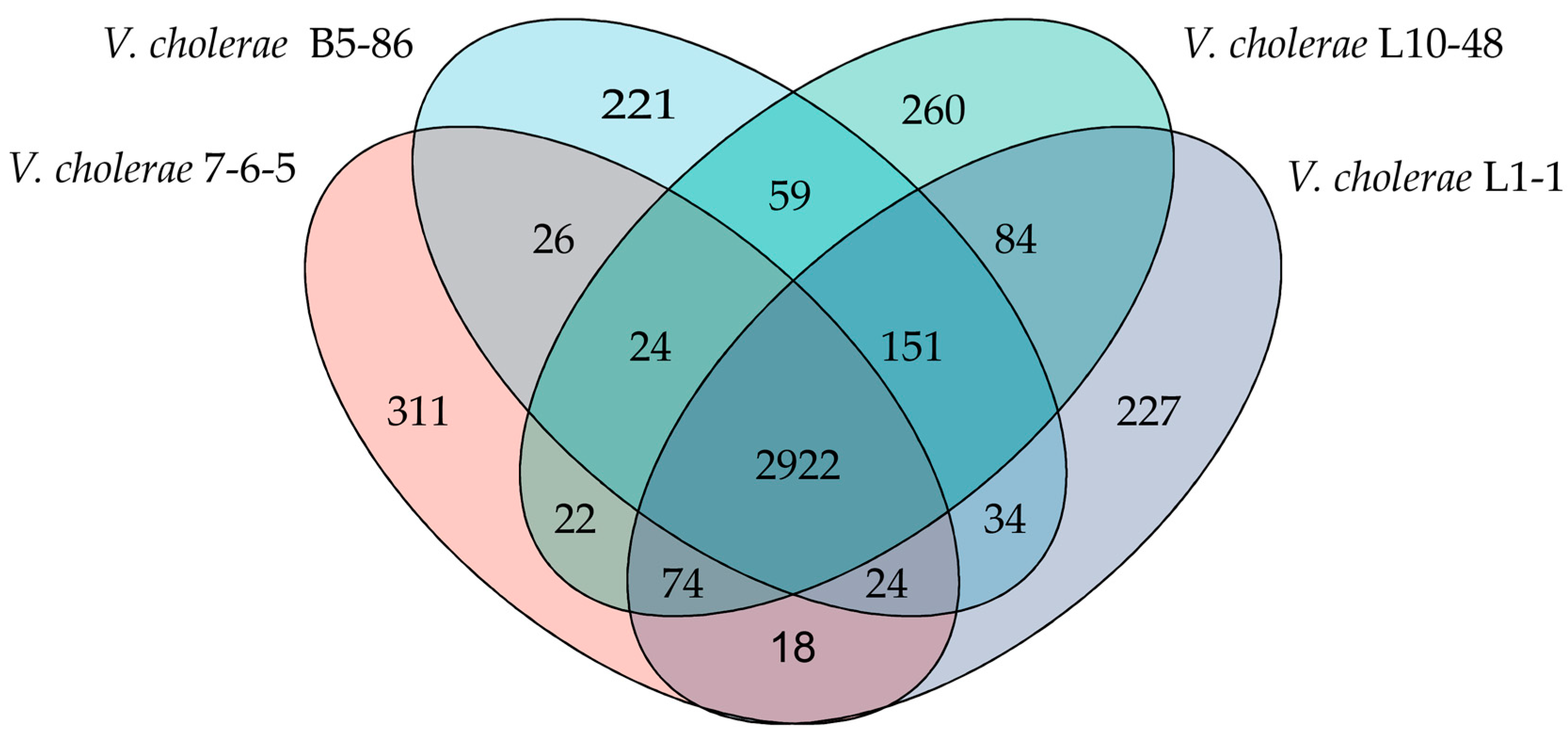
3.7. Strain-Specific Genes of the Four V. cholerae Isolates from the Common Freshwater Fish
3.8. MLST of the Four V. cholerae Isolates from the Common Freshwater Fish
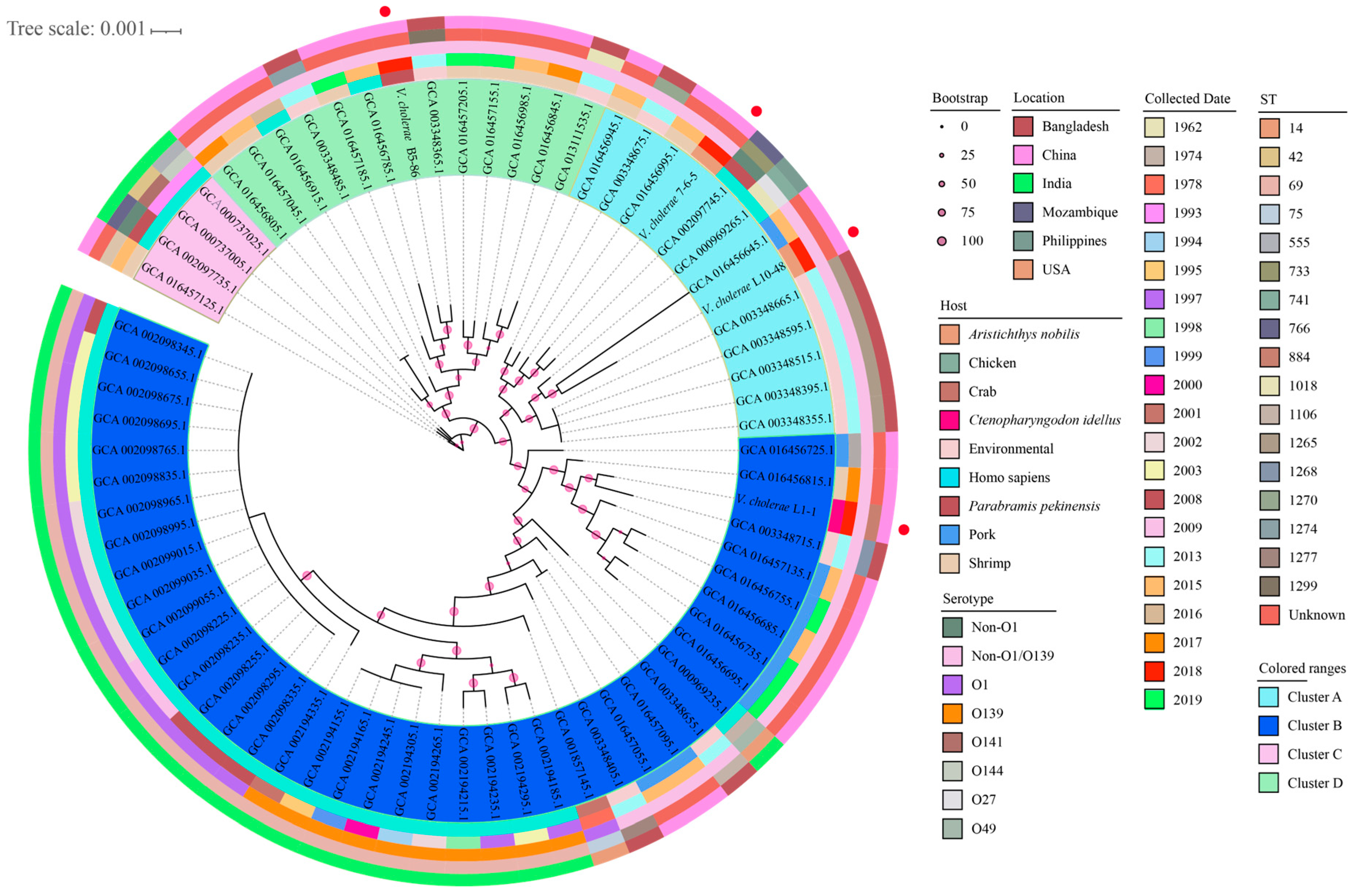
3.9. Phylogenetic Relatedness of the Four V. cholerae Isolates from the Common Freshwater Fish
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Montero, D.A.; Vidal, R.M.; Velasco, J.; George, S.; Lucero, Y.; Gómez, L.A.; Carreño, L.J.; García-Betancourt, R.; O’Ryan, M. Vibrio cholerae, classification, pathogenesis, immune response, and trends in vaccine development. Front. Med. 2023, 10, 1155751. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Wu, J.; Chen, L. Virulence, antimicrobial and heavy metal tolerance, and genetic diversity of Vibrio cholerae recovered from commonly consumed freshwater fish. Environ. Sci. Pollut. Res. Int. 2019, 26, 27338–27352. [Google Scholar] [CrossRef] [PubMed]
- Waldor, M.K.; Mekalanos, J.J. Lysogenic conversion by a filamentous phage encoding cholera toxin. Science 1996, 272, 1910–1914. [Google Scholar] [CrossRef] [PubMed]
- Almansour, A.M.; Alhadlaq, M.A.; Alzahrani, K.O.; Mukhtar, L.E.; Alharbi, A.L.; Alajel, S.M. The silent threat: Antimicrobial-resistant pathogens in food-producing animals and their impact on public health. Microorganisms 2023, 11, 2127. [Google Scholar] [CrossRef]
- Taviani, E.; van den Berg, H.; Nhassengo, F.; Nguluve, E.; Paulo, J.; Pedro, O.; Ferrero, G. Occurrence of waterborne pathogens and antibiotic resistance in water supply systems in a small town in Mozambique. BMC Microbiol. 2022, 22, 243. [Google Scholar] [CrossRef] [PubMed]
- Abioye, O.E.; Nontongana, N.; Osunla, C.A.; Okoh, A.I. Antibiotic resistance and virulence genes profiling of Vibrio cholerae and Vibrio mimicus isolates from some seafood collected at the aquatic environment and wet markets in Eastern Cape Province, South Africa. PLoS ONE 2023, 18, e0290356. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, K.; Scholz, H.C.; Appelt, S.; Michel, J.; Jacob, D.; Dupke, S. Virulence and resistance patterns of Vibrio cholerae non-O1/non-O139 acquired in Germany and other European countries. Front. Microbiol. 2023, 14, 1282135. [Google Scholar] [CrossRef] [PubMed]
- Arnold, B.J.; Huang, I.T.; Hanage, W.P. Horizontal gene transfer and adaptive evolution in bacteria. Nat. Rev. Microbiol. 2022, 20, 206–218. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Gu, T.; Ou, Y.; Wang, Y.; Xie, L.; Chen, L. Environmental compatibility and genome flexibility of Klebsiella oxytoca isolated from eight species of aquatic animals. Diversity 2024, 16, 30. [Google Scholar] [CrossRef]
- Haudiquet, M.; Sousa, J.M.; Touchon, M.; Rocha, E.P.C. Selfish, promiscuous and sometimes useful: How mobile genetic elements drive horizontal gene transfer in microbial populations. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2022, 377, 20210234. [Google Scholar] [CrossRef]
- Dorado, G.; Gálvez, S.; Rosales, T.E.; Vásquez, V.F.; Hernández, P. Analyzing modern biomolecules: The revolution of nucleic-acid sequencing—Review. Biomolecules 2021, 11, 1111. [Google Scholar] [CrossRef] [PubMed]
- Evrony, G.D.; Hinch, A.G.; Luo, C. Applications of single-cell DNA sequencing. Annu. Rev. Genom. Hum. G. 2021, 22, 171–197. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Li, X.; Ni, L.; Xu, D.; Xu, Y.; Ding, Y.; Xie, L.; Chen, L. First experimental evidence for the presence of potentially toxic Vibrio cholerae in snails, and virulence, cross-resistance and genetic diversity of the bacterium in 36 species of aquatic food animals. Antibiotics 2021, 10, 412. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.; Yu, P.; Liang, W.; Kan, B.; Peng, X.; Chen, L. Virulence, resistance, and genomic fingerprint traits of Vibrio cholerae isolated from 12 species of aquatic products in Shanghai, China. Microb. Drug. Resist. 2020, 26, 1526–1539. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Yu, P.; Li, B.; Pan, Y.; Zhang, X.; Cong, J.; Zhao, Y.; Wang, H.; Chen, L. The mosaic accessory gene structures of the SXT/R391-like integrative and conjugative elements derived from Vibrio spp. isolated from aquatic products and environment in the Yangtze River Estuary, China. BMC Microbiol. 2013, 13, 214. [Google Scholar] [CrossRef]
- Miller, C.J.; Drasar, B.S.; Feachem, R.G. Response of toxigenic Vibrio cholerae 01 to physico-chemical stresses in aquatic environments. J. Hygiene. 1984, 93, 475–495. [Google Scholar] [CrossRef] [PubMed]
- Guan, H.; Xie, L.; Chen, L. Survival and genome evolution signatures of Klebsiella pneumoniae isolates originated in seven species of aquatic animals. Diversity 2023, 15, 527. [Google Scholar] [CrossRef]
- Xu, D.; Peng, X.; Xie, L.; Chen, L. Survival and genome diversity of Vibrio parahaemolyticus isolated from edible aquatic animals. Diversity 2022, 14, 350. [Google Scholar] [CrossRef]
- Yu, P.; Yang, L.; Wang, J.; Su, C.; Qin, S.; Zeng, C.; Chen, L. Genomic and transcriptomic analysis reveal multiple strategies for the cadmium tolerance in Vibrio parahaemolyticus N10-18 isolated from aquatic animal Ostrea Gigas Thunberg. Foods 2022, 11, 3777. [Google Scholar] [CrossRef] [PubMed]
- Luo, R.; Liu, B.; Xie, Y.; Li, Z.; Huang, W.; Yuan, J.; He, G.; Chen, Y.; Pan, Q.; Liu, Y.; et al. SOAPdenovo2: An empirically improved memory-efficient short-read de novo assembler. GigaScience 2012, 1, 18. [Google Scholar] [CrossRef]
- Delcher, A.L.; Bratke, K.A.; Powers, E.C.; Salzberg, S.L. Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics 2007, 23, 673–679. [Google Scholar] [CrossRef] [PubMed]
- Chan, P.P.; Lowe, T.M. tRNAscan-SE: Searching for tRNA genes in genomic sequences. Methods Mol. Biol. 2019, 1962, 1–14. [Google Scholar] [CrossRef]
- Pal, C.; Bengtsson-Palme, J.; Rensing, C.; Kristiansson, E.; Larsson, D.G.J. BacMet: Antibacterial biocide and metal resistance genes database. Nucleic. Acids. Res. 2014, 42, D737–D743. [Google Scholar] [CrossRef] [PubMed]
- Bertelli, C.; Fiona, S.L. Improved genomic island predictions with IslandPath-DIMOB. Bioinformatics 2018, 34, 2161–2167. [Google Scholar] [CrossRef]
- Fouts, D.E. Phage_Finder: Automated identification and classification of prophage regions in complete bacterial genome sequences. Nucleic. Acids. Res. 2006, 34, 5839–5851. [Google Scholar] [CrossRef]
- Cury, J.; Jové, T.; Touchon, M.; Néron, B.; Rocha, E.P. Identification and analysis of integrons and cassette arrays in bacterial genomes. Nucleic. Acids. Res. 2016, 44, 4539–4550. [Google Scholar] [CrossRef]
- Siguier, P.; Perochon, J.; Lestrade, L.; Mahillon, J.; Chandler, M. ISfinder: The reference centre for bacterial insertion sequences. Nucleic. Acids. Res. 2006, 34, D32–D36. [Google Scholar] [CrossRef]
- Bland, C.; Ramsey, T.L.; Sabree, F.; Lowe, M.; Brown, K.; Kyrpides, N.C.; Hugenholtz, P. CRISPR recognition tool (CRT): A tool for automatic detection of clustered regularly interspaced palindromic repeats. BMC Bioinform. 2007, 8, 209. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Stoeckert, C.J., Jr.; Roos, D.S. OrthoMCL: Identification of ortholog groups for eukaryotic genomes. Genome. Res. 2003, 13, 2178–2189. [Google Scholar] [CrossRef]
- Jolley, K.A.; Bray, J.E.; Maiden, M.C.J. Open-access bacterial population genomics: BIGSdb software, the PubMLST.org website and their applications. Wellcome. Open. Res. 2018, 3, 124. [Google Scholar] [CrossRef]
- Heidelberg, J.F.; Eisen, J.A.; Nelson, W.C.; Clayton, R.A.; Gwinn, M.L.; Dodson, R.J.; Haft, D.H.; Hickey, E.K.; Peterson, J.D.; Umayam, L.; et al. DNA sequence of both chromosomes of the cholera pathogen Vibrio cholerae. Nature 2000, 406, 477–483. [Google Scholar] [CrossRef] [PubMed]
- McKerral, J.C.; Papudeshi, B.; Inglis, L.K.; Roach, M.J.; Decewicz, P.; McNair, K.; Luque, A.; Dinsdale, E.A.; Edwards, R.A. The promise and pitfalls of prophages. bioRxiv 2023. bioRxiv: 537752. [Google Scholar] [CrossRef] [PubMed]
- An, X.L.; Chen, Q.L.; Zhu, D.; Zhu, Y.G.; Gillings, M.R.; Su, J.Q. Impact of wastewater treatment on the prevalence of integrons and the genetic diversity of integron gene gassettes. Appl. Environ. Microb. 2018, 84, e02766-17. [Google Scholar] [CrossRef] [PubMed]
- Sabbagh, P.; Rajabnia, M.; Maali, A.; Ferdosi-Shahandashti, E. Integron and its role in antimicrobial resistance: A literature review on some bacterial pathogens. Iran. J. Basic. Med. Sci. 2021, 24, 136–142. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, O.S.; Campos, K.F.; Assis, J.C.S.; Fernandes, A.S.; Souza, T.S.; Rodrigues, L.G.C.; Queiroz, M.V.; Santana, M.F. Transposable elements contribute to the genome plasticity of Ralstonia solanacearum species complex. Microb. Genom. 2020, 6, e000374. [Google Scholar] [CrossRef]
- Zakrzewska, M.; Burmistrz, M. Mechanisms regulating the CRISPR-Cas systems. Front. Microbiol. 2023, 14, 1060337. [Google Scholar] [CrossRef] [PubMed]
- Deveau, H.; Garneau, J.E.; Moineau, S. CRISPR/Cas system and its role in phage-bacteria interactions. Annu. Rev. Microbiol. 2010, 64, 475–493. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Du, X.; Zhao, Y.; Wang, W.; Cai, T.; Tang, K.; Wang, X. Combining CRISPR/Cas9 and natural excision for the precise and complete removal of mobile genetic elements in bacteria. Appl. Environ. Microbiol. 2024, 90, e0009524. [Google Scholar] [CrossRef]
- Austin, B. Vibrios as causal agents of zoonoses. Vet. Microbiol. 2010, 140, 310–317. [Google Scholar] [CrossRef]
- Ceccarelli, D.; Chen, A.; Hasan, N.A.; Rashed, S.M.; Huq, A.; Colwell, R.R. Non-O1/non-O139 Vibrio cholerae carrying multiple virulence factors and V. cholerae O1 in the Chesapeake Bay, Maryland. Appl. Environ. Microbiol. 2015, 81, 1909–1918. [Google Scholar] [CrossRef]
- Lee, A.; White, N.; Walle, C.F. The intestinal zonula occludens toxin (ZOT) receptor recognises non-native ZOT conformers and localises to the intercellular contacts. FEBS Lett. 2003, 555, 638–642. [Google Scholar] [CrossRef] [PubMed]
- Dong, T.G.; Ho, B.T.; Yoder-Himes, D.R.; Mekalanos, J.J. Identification of T6SS-dependent effector and immunity proteins by Tn-seq in Vibrio cholerae. Proc. Natl. Acad. Sci. USA 2013, 110, 2623–2628. [Google Scholar] [CrossRef] [PubMed]
- Kodama, T.; Rokuda, M.; Park, K.S.; Cantarelli, V.V.; Matsuda, S.; Iida, T.; Honda, T. Identification and characterization of VopT, a novel ADP-ribosyltransferase effector protein secreted via the Vibrio parahaemolyticus type III secretion system 2. Cell. Microbiol. 2007, 9, 2598–2609. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Wang, X.; Li, J.; Xue, F.; Tang, F.; Dai, J. Extraintestinal pathogenic Escherichia coli utilizes the surface-expressed elongation factor Tu to bind and acquire iron from holo-transferrin. Virulence 2022, 13, 698–713. [Google Scholar] [CrossRef] [PubMed]
- Inzana, T.J. The many facets of lipooligosaccharide as a virulence factor for Histophilus somni. Curr. Top. Microbiol. Immunol. 2016, 396, 131–148. [Google Scholar] [PubMed]
- Sheahan, K.L.; Cordero, C.L.; Satchell, K.J. Identification of a domain within the multifunctional Vibrio cholerae RTX toxin that covalently cross-links actin. Proc. Natl. Acad. Sci. USA 2004, 101, 9798–9803. [Google Scholar] [CrossRef] [PubMed]
- De, S.; Olson, R. Crystal structure of the Vibrio cholerae cytolysin heptamer reveals common features among disparate pore-forming toxins. Proc. Natl. Acad. Sci. USA 2011, 108, 7385–7390. [Google Scholar] [CrossRef]
- Nishino, K.; Senda, Y.; Yamaguchi, A. CRP regulator modulates multidrug resistance of Escherichia coli by repressing the mdtEF multidrug efflux genes. J. Antibiot. 2008, 61, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Henderson, J.C.; Herrera, C.M.; Trent, M.S. AlmG, responsible for polymyxin resistance in pandemic Vibrio cholerae, is a glycyltransferase distantly related to lipid A late acyltransferases. J. Biol. Chem. 2017, 292, 21205–21215. [Google Scholar] [CrossRef]
- Tacão, M.; Moura, A.; Correia, A.; Henriques, I. Co-resistance to different classes of antibiotics among ESBL-producers from aquatic systems. Water Res. 2014, 48, 100–107. [Google Scholar] [CrossRef]
- Lin, H.V.; Massam-Wu, T.; Lin, C.P.; Wang, Y.A.; Shen, Y.; Lu, W.; Hsu, P.; Chen, Y.; Borges-Walmsley, M.I.; Walmsley, A.R. The Vibrio cholerae var regulon encodes a metallo-β-lactamase and an antibiotic efflux pump, which are regulated by VarR, a LysR-type transcription factor. PLoS ONE 2017, 12, e0184255. [Google Scholar] [CrossRef] [PubMed]
- Lepuschitz, S.; Baron, S.; Larvor, E.; Granier, S.A.; Pretzer, C.; Mach, R.L.; Farnleitner, A.H.; Ruppitsch, W.; Pleininger, S.; Indra, A.; et al. Phenotypic and genotypic antimicrobial resistance traits of Vibrio cholerae non-O1/non-O139 isolated from a large Austrian lake frequently associated with cases of human infection. Front. Microbiol. 2019, 10, 2600. [Google Scholar] [CrossRef]
- Melano, R.; Petroni, A.; Garutti, A.; Saka, H.A.; Mange, L.; Pasterán, F.; Rapoport, M.; Rossi, A.; Galas, M. New carbenicillin-hydrolyzing β-lactamase (CARB-7) from Vibrio cholerae non-O1, non-O139 strains encoded by the VCR region of the V. cholerae genome. Antimicrob. Agents Chemother. 2002, 46, 2162–2168. [Google Scholar] [CrossRef] [PubMed]
- Lacour, S.; Bechet, E.; Cozzone, A.J.; Mijakovic, I.; Grangeasse, C. Tyrosine phosphorylation of the UDP-glucose dehydrogenase of Escherichia coli is at the crossroads of colanic acid synthesis and polymyxin resistance. PLoS ONE 2008, 3, e3053. [Google Scholar] [CrossRef] [PubMed]
- Bonifer, C.; Glaubitz, C. MsbA: An ABC transporter paradigm. Biochem. Soc. Trans. 2021, 49, 2917–2927. [Google Scholar] [CrossRef] [PubMed]
- Siddi, G.; Piras, F.; Meloni, M.P.; Gymoese, P.; Torpdahl, M.; Fredriksson-Ahomaa, M.; Migoni, M.; Cabras, D.; Cuccu, M.; De Santis, E.P.L.; et al. Hunted wild boars in sardinia: Prevalence, antimicrobial resistance and genomic analysis of Salmonella and Yersinia enterocolitica. Foods 2023, 13, 65. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.H.T.; Nguyen, H.D.; Le, M.H.; Nguyen, T.T.H.; Nguyen, T.D.; Nguyen, D.L.; Nguyen, Q.H.; Nguyen, T.K.O.; Michalet, S.; Dijoux-Franca, M.; et al. Efflux pump inhibitors in controlling antibiotic resistance: Outlook under a heavy metal contamination context. Molecules 2023, 28, 2912. [Google Scholar] [CrossRef] [PubMed]
- Syed, K.A.; Beyhan, S.; Correa, N.; Queen, J.; Liu, J.; Peng, F.; Satchell, K.J.F. Yildiz, F.; Klose, K.E. The Vibrio cholerae flagellar regulatory hierarchy controls expression of virulence factors. J. Bacteriol. 2009, 191, 6555–6570. [Google Scholar] [CrossRef] [PubMed]
- Utada, A.S.; Bennett, R.R.; Fong, J.C.N.; Gibiansky, M.L.; Yildiz, F.H.; Golestanian, R.; Wong, G.C.L. Vibrio cholerae use pili and flagella synergistically to effect motility switching and conditional surface attachment. Nat. Commun. 2014, 5, 4913. [Google Scholar] [CrossRef]
- Chen, X.; Schauder, S.; Potier, N.; Dorsselaer, A.V.; Pelczer, I.; Bassler, B.L.; Hughson, F.M. Structural identification of a bacterial quorum-sensing signal containing boron. Nature. 2002, 415, 545–549. [Google Scholar] [CrossRef]
- Chen, L.; Yang, J.; Yu, J.; Yao, Z.; Sun, L.; Shen, Y.; Jin, Q. VFDB: A reference database for bacterial virulence factors. Nucleic. Acids. Res. 2005, 33, D325–D328. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.Y.; Lee, H.Y.; Lee, K.H.; Han, S.H.; Park, S.J. Vibrio vulnificus IlpA induces MAPK-mediated cytokine production via TLR1/2 activation in THP-1 cells, a human monocytic cell line. Mol. Immunol. 2011, 49, 143–154. [Google Scholar] [CrossRef] [PubMed]
- Fullner, K.J.; Mekalanos, J.J. In vivo covalent cross-linking of cellular actin by the Vibrio cholerae RTX toxin. EMBO J. 2000, 19, 5315–5323. [Google Scholar] [CrossRef]
- Higgins, D.A.; Pomianek, M.E.; Kraml, C.M.; Taylor, R.K.; Semmelhack, M.F.; Bassler, B.L. The major Vibrio cholerae autoinducer and its role in virulence factor production. Nature 2007, 450, 883–886. [Google Scholar] [CrossRef] [PubMed]
| Genome Feature | V. cholerae Isolate | |||
|---|---|---|---|---|
| 7-6-5 | L10-48 | L1-1 | B5-86 | |
| Genome size (bp) | 3,886,208 | 4,145,566 | 4,032,220 | 3,942,938 |
| G + C (%) | 47.44 | 47.35 | 47.58 | 47.63 |
| DNA Scaffold | 175 | 80 | 79 | 49 |
| Predicted gene | 3500 | 3663 | 3570 | 3487 |
| Protein coding gene | 3404 | 3561 | 3476 | 3366 |
| RNA gene | 96 | 102 | 94 | 121 |
| Genes assigned to COG | 2981 | 3046 | 3010 | 2985 |
| Genes with unknown function | 519 | 617 | 560 | 502 |
| GIs | 4 | 9 | 6 | 4 |
| Prophage gene cluster | 0 | 2 | 3 | 1 |
| Ins | 1 | 1 | 1 | 1 |
| ISs | 0 | 3 | 1 | 2 |
| CRISPR-Cas array | 8 | 10 | 6 | 2 |
| BioSample accession no. | SAMN37882014 | SAMN37882015 | SAMN37882016 | SAMN37882017 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qin, X.; Yang, L.; Xu, Y.; Xie, L.; Wang, Y.; Chen, L. Growth and Genome Features of Non-O1/O139 Vibrio cholerae Isolated from Three Species of Common Freshwater Fish. Diversity 2024, 16, 268. https://doi.org/10.3390/d16050268
Qin X, Yang L, Xu Y, Xie L, Wang Y, Chen L. Growth and Genome Features of Non-O1/O139 Vibrio cholerae Isolated from Three Species of Common Freshwater Fish. Diversity. 2024; 16(5):268. https://doi.org/10.3390/d16050268
Chicago/Turabian StyleQin, Xinchi, Lianzhi Yang, Yingwei Xu, Lu Xie, Yongjie Wang, and Lanming Chen. 2024. "Growth and Genome Features of Non-O1/O139 Vibrio cholerae Isolated from Three Species of Common Freshwater Fish" Diversity 16, no. 5: 268. https://doi.org/10.3390/d16050268
APA StyleQin, X., Yang, L., Xu, Y., Xie, L., Wang, Y., & Chen, L. (2024). Growth and Genome Features of Non-O1/O139 Vibrio cholerae Isolated from Three Species of Common Freshwater Fish. Diversity, 16(5), 268. https://doi.org/10.3390/d16050268