
Genomic Patterns of Iberian Wild Bees Reveal Levels of Diversity, Differentiation and Population Structure, Supporting the “Refugia within Refugia” Hypothesis

 , , ,
, , ,  , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
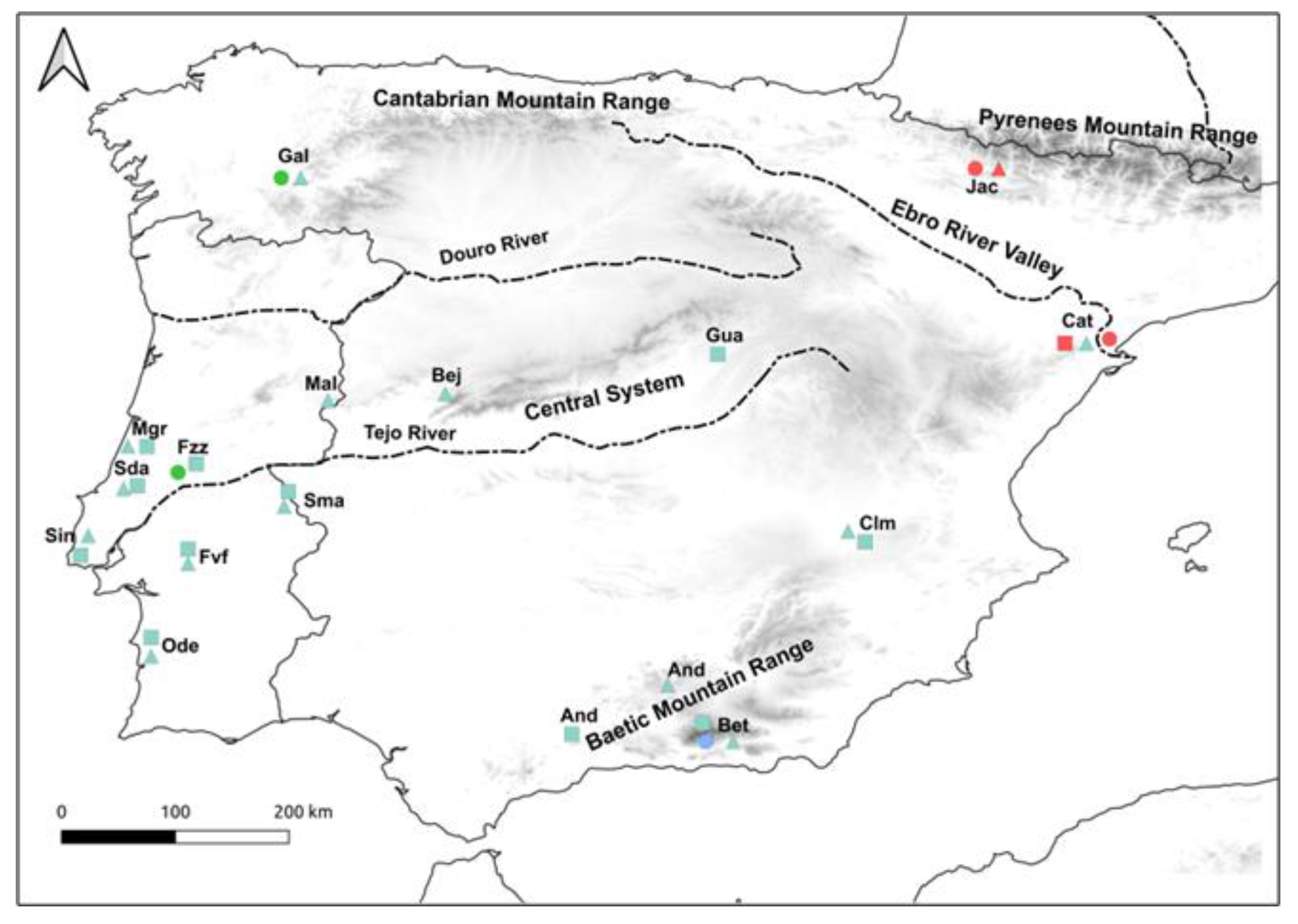
2.1. Sampling
2.2. DNA Extraction
2.3. COI Amplification
2.4. SNP Calling and Filtering
2.5. Population Structure
2.6. Summary Statistics
3. Results
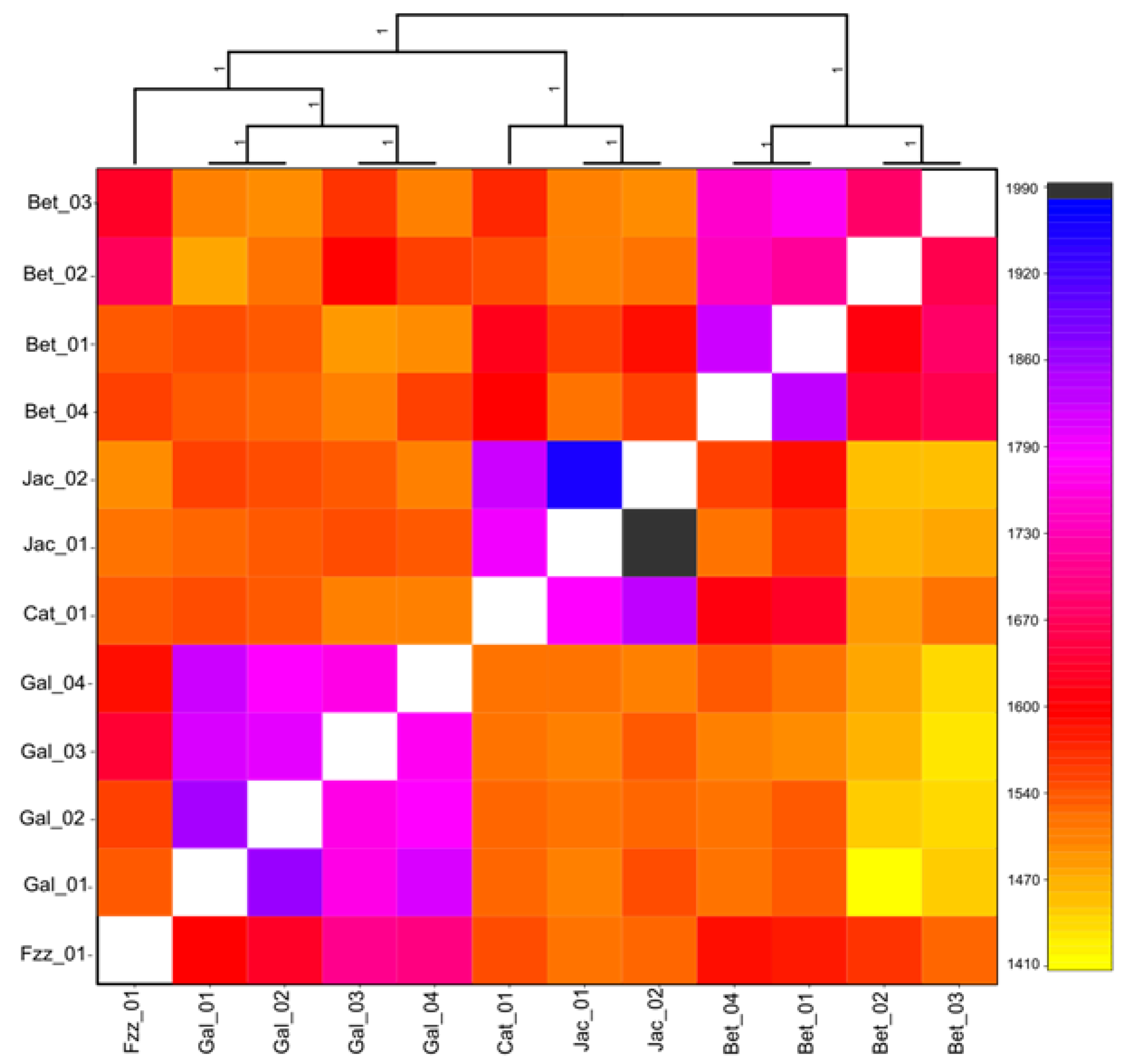
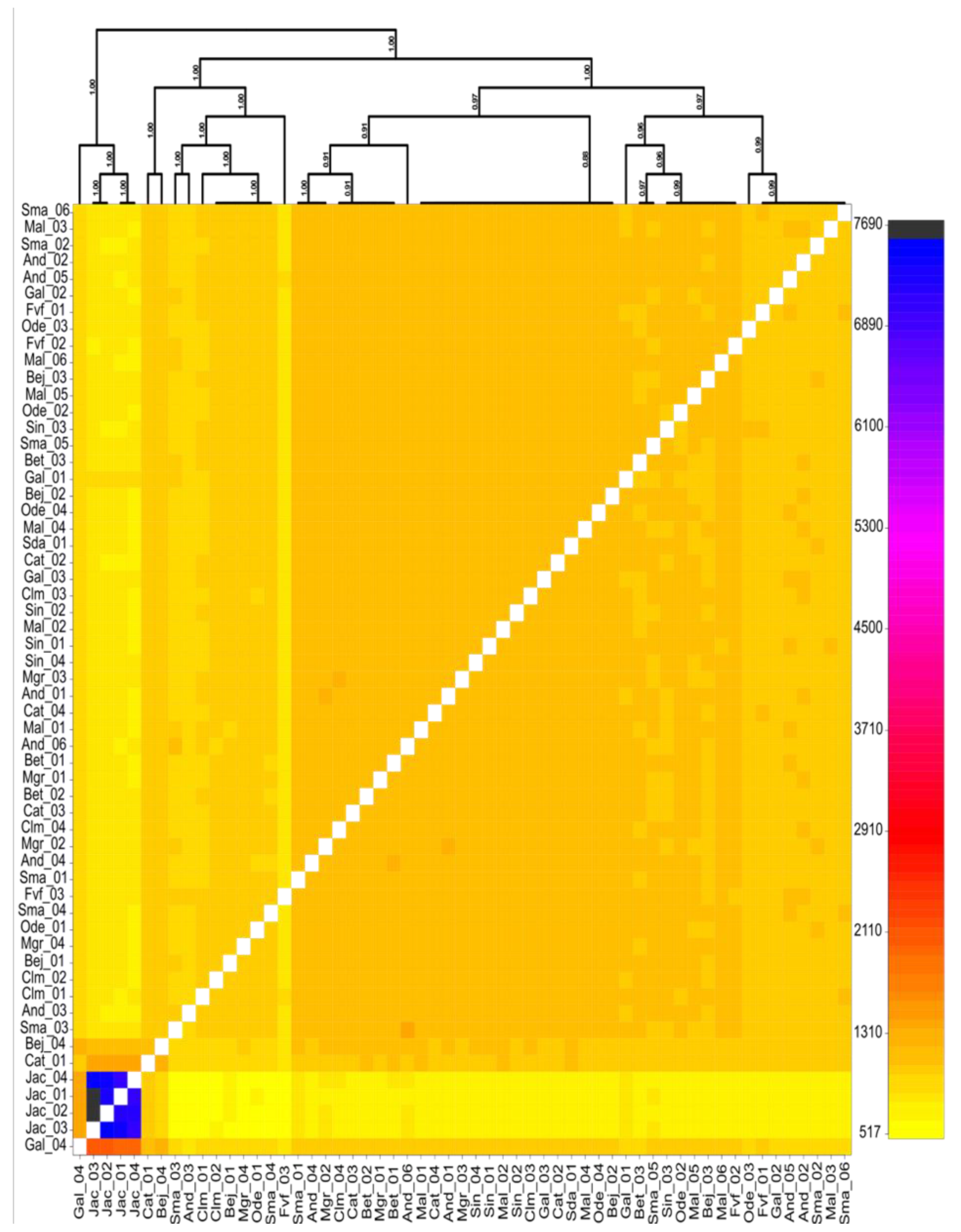
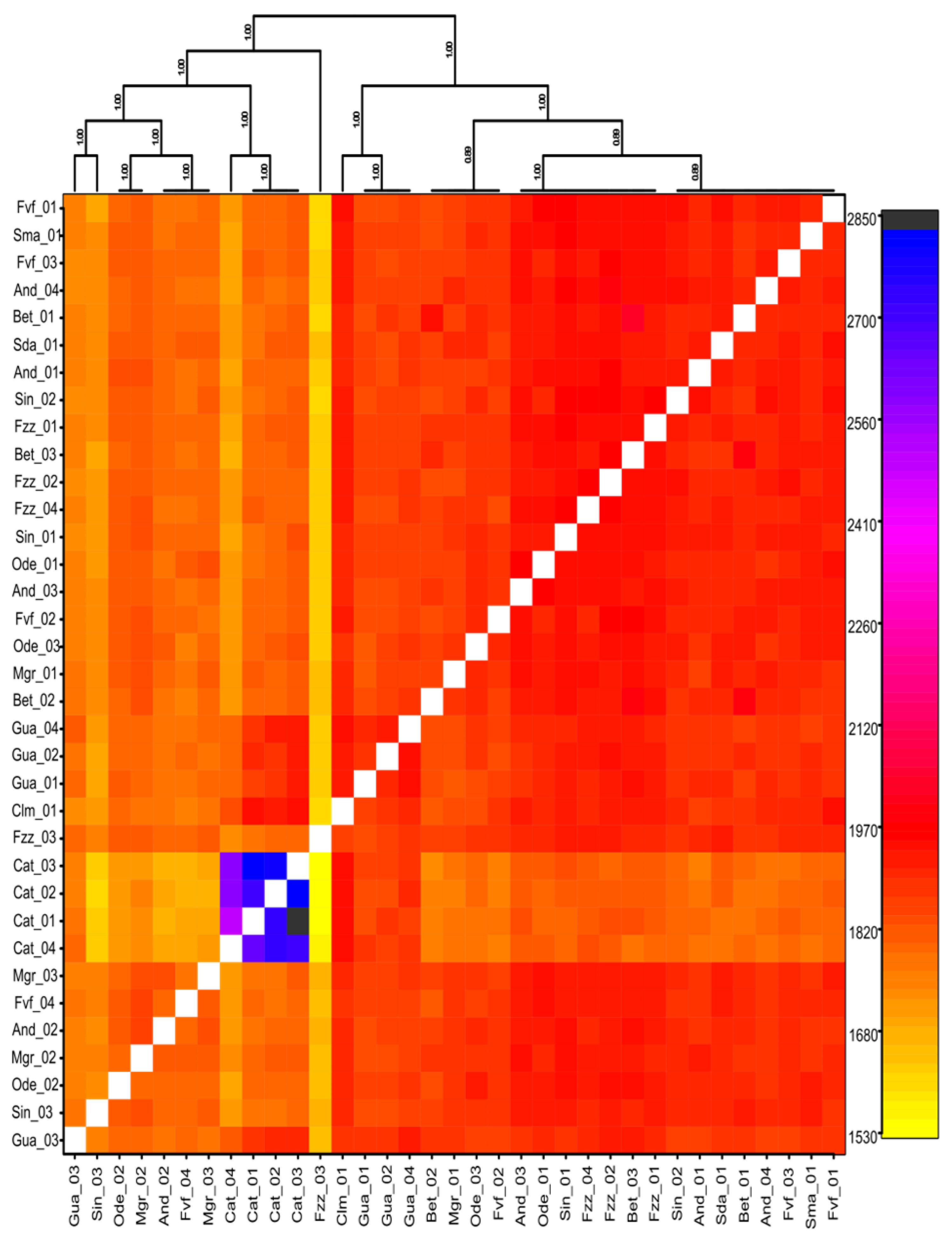
3.1. Population Structure
3.2. Summary Statistics
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Cardoso, P.; Barton, P.S.; Birkhofer, K.; Chichorro, F.; Deacon, C.; Fartmann, T.; Fukushima, C.S.; Gaigher, R.; Habel, J.C.; Hallmann, C.A.; et al. Scientists’ Warning to Humanity on Insect Extinctions. Biol. Conserv. 2020, 242, 108426. [Google Scholar] [CrossRef]
- Potts, S.G.; Biesmeijer, J.C.; Kremen, C.; Neumann, P.; Schweiger, O.; Kunin, W.E. Global Pollinator Declines: Trends, Impacts and Drivers. Trends Ecol. Evol. 2010, 25, 345–353. [Google Scholar] [CrossRef]
- Ollerton, J.; Winfree, R.; Tarrant, S. How Many Flowering Plants Are Pollinated by Animals? Oikos 2011, 120, 321–326. [Google Scholar] [CrossRef]
- Bates, A.J.; Sadler, J.P.; Fairbrass, A.J.; Falk, S.J.; Hale, J.D.; Matthews, T.J. Changing Bee and Hoverfly Pollinator Assemblages along an Urban-Rural Gradient. PLoS ONE 2011, 6, e23459. [Google Scholar] [CrossRef] [PubMed]
- Winfree, R.; Bartomeus, I.; Cariveau, D.P. Native Pollinators in Anthropogenic Habitats. Annu. Rev. Ecol. Evol. Syst. 2011, 42, 1–22. [Google Scholar] [CrossRef]
- Nieto, A.; Roberts, S.P.M.; Kemp, J.; Rasmont, P.; Kuhlmann, M.; García Criado, M.; Biesmeijer, J.C.; Bogusch, P.; Dathe, H.H.; De la Rúa, P.; et al. European Red List of Bees; Publication Office of the European Union: Luxembourg, 2014; ISBN 9789279445125. [Google Scholar]
- Danforth, B.N.; Cardinal, S.; Praz, C.; Almeida, E.A.B.; Michez, D. The Impact of Molecular Data on Our Understanding of Bee Phylogeny and Evolution. Annu. Rev. Entomol. 2013, 58, 57–78. [Google Scholar] [CrossRef]
- Geldmann, J.; González-Varo, J.P. Conserving Honey Bees Does Not Help Wildlife. Science 2018, 359, 392–393. [Google Scholar] [CrossRef]
- Wood, T.J.; Michez, D.; Paxton, R.J.; Drossart, M.; Neumann, P.; Gérard, M.; Vanderplanck, M.; Barraud, A.; Martinet, B.; Leclercq, N.; et al. Managed Honey Bees as a Radar for Wild Bee Decline? Apidologie 2020, 51, 1100–1116. [Google Scholar] [CrossRef]
- López-Uribe, M.M.; Ricigliano, V.A.; Simone-Finstrom, M. Defining Pollinator Health: A Holistic Approach Based on Ecological, Genetic, and Physiological Factors. Annu. Rev. Anim. Biosci. 2020, 8, 269–294. [Google Scholar] [CrossRef]
- Pauls, S.U.; Nowak, C.; Balint, M.; Pfenninger, M. The Impact of Global Climate Change on Genetic Diversity within Populations and Species. Mol. Ecol. 2013, 22, 925–946. [Google Scholar] [CrossRef]
- Maebe, K.; Karise, R.; Meeus, I.; Mänd, M.; Smagghe, G. Pattern of Population Structuring between Belgian and Estonian Bumblebees. Sci. Rep. 2019, 9, 9651. [Google Scholar] [CrossRef]
- Maebe, K.; Meeus, I.; Ganne, M.; De Meulemeester, T.; Biesmeijer, K.; Smagghe, G. Microsatellite Analysis of Museum Specimens Reveals Historical Differences in Genetic Diversity between Declining and More Stable Bombus Species. PLoS ONE 2015, 10, e0127870. [Google Scholar] [CrossRef]
- Maebe, K.; Meeus, I.; Vray, S.; Claeys, T.; Dekoninck, W.; Boevé, J.L.; Rasmont, P.; Smagghe, G. A Century of Temporal Stability of Genetic Diversity in Wild Bumblebees. Sci. Rep. 2016, 6, 38289. [Google Scholar] [CrossRef]
- Dreier, S.; Redhead, J.W.; Warren, I.A.; Bourke, A.F.G.; Heard, M.S.; Jordan, W.C.; Sumner, S.; Wang, J.; Carvell, C. Fine-Scale Spatial Genetic Structure of Common and Declining Bumble Bees across an Agricultural Landscape. Mol. Ecol. 2014, 23, 3384–3395. [Google Scholar] [CrossRef]
- Lozier, J.D. Revisiting Comparisons of Genetic Diversity in Stable and Declining Species: Assessing Genome-Wide Polymorphism in North American Bumble Bees Using RAD Sequencing. Mol. Ecol. 2014, 23, 788–801. [Google Scholar] [CrossRef]
- Orr, M.C.; Hughes, A.C.; Chesters, D.; Pickering, J.; Zhu, C.D.; Ascher, J.S. Global Patterns and Drivers of Bee Distribution. Curr. Biol. 2021, 31, 451–458.e4. [Google Scholar] [CrossRef]
- Taberlet, P.; Fumagalli, L.; Wust-Saucy, A.G.; Cosson, J.F. Comparative Phylogeography and Postglacial Colonization Routes in Europe. Mol. Ecol. 1998, 7, 453–464. [Google Scholar] [CrossRef]
- Hewitt, G. The Genetic Legacy of the Quaternary Ice Ages. Nature 2000, 405, 907–913. [Google Scholar] [CrossRef]
- Gómez, A.; Lunt, D.H. Refugia within Refugia: Patterns of Phylogeographic Concordance in the Iberian Peninsula. In Phylogeography of Southern European Refugia; Weiss, S., Ferrand, N., Eds.; Springer: Berlin/Heidelberg, Germany, 2007; pp. 155–188. ISBN 9781402049033. [Google Scholar]
- Blondel, J.; Aronson, J.; Bodiou, J.-Y.; Boeuf, G. The Mediterranean Region Biological Diversity in Space and Time, 2nd ed.; Oxford University Press: Oxford, UK, 2010. [Google Scholar]
- Miraldo, A.; Hewitt, G.M.; Paulo, O.S.; Emerson, B.C. Phylogeography and Demographic History of Lacerta Lepida in the Iberian Peninsula: Multiple Refugia, Range Expansions and Secondary Contact Zones. BMC Evol. Biol. 2011, 11, 170. [Google Scholar] [CrossRef]
- Chávez-Galarza, J.; Henriques, D.; Johnston, J.S.; Carneiro, M.; Rufino, J.; Patton, J.C.; Pinto, M.A. Revisiting the Iberian Honey Bee (Apis mellifera iberiensis) Contact Zone: Maternal and Genome-Wide Nuclear Variations Provide Support for Secondary Contact from Historical Refugia. Mol. Ecol. 2015, 24, 2973–2992. [Google Scholar] [CrossRef]
- Silva, S.E.; Seabra, S.G.; Carvalheiro, L.G.; Nunes, V.L.; Marabuto, E.; Mendes, R.; Rodrigues, A.S.B.; Pina-Martins, F.; Yurtsever, S.; Laurentino, T.G.; et al. Population Genomics of Bombus Terrestris Reveals High but Unstructured Genetic Diversity in a Potential Glacial Refugium. Biol. J. Linn. Soc. 2020, 129, 259–272. [Google Scholar] [CrossRef]
- Hewitt, G.M. Some Genetic Consequences of Ice Ages, and Their Role, in Divergence and Speciation. Biol. J. Linn. Soc. 1996, 58, 247–276. [Google Scholar] [CrossRef]
- Packer, L.; Zayed, A.; Grixti, J.C.; Ruz, L.; Owen, R.E.; Vivallo, F.; Toro, H. Conservation Genetics of Potentially Endangered Mutualisms: Reduced Levels of Genetic Variation in Specialist versus Generalist Bees. Conserv. Biol. 2005, 19, 195–202. [Google Scholar] [CrossRef]
- Dellicour, S.; Michez, D.; Mardulyn, P. Comparative Phylogeography of Five Bumblebees: Impact of Range Fragmentation, Range Size and Diet Specialization. Biol. J. Linn. Soc. 2015, 116, 926–939. [Google Scholar] [CrossRef]
- Dellicour, S.; Michez, D.; Rasplus, J.-Y.; Mardulyn, P. Impact of Past Climatic Changes and Resource Availability on the Population Demography of Three Food-Specialist Bees. Mol. Ecol. 2015, 24, 1074–1090. [Google Scholar] [CrossRef]
- Lepais, O.; Darvill, B.; O’Connor, S.; Osborne, J.L.; Sanderson, R.A.; Cussans, J.; Goffe, L.; Goulson, D. Estimation of Bumblebee Queen Dispersal Distances Using Sibship Reconstruction Method. Mol. Ecol. 2010, 19, 819–831. [Google Scholar] [CrossRef]
- López-Uribe, M.M.; Jha, S.; Soro, A. A Trait-Based Approach to Predict Population Genetic Structure in Bees. Mol. Ecol. 2019, 28, 1–11. [Google Scholar] [CrossRef]
- Falk, S.J. Field Guide to the Bees of Great Britain and Ireland; Bloomsbury Publishing: London, UK, 2015; Volume 14, ISBN 9781472967053. [Google Scholar]
- Güler, Y.; Sorkun, K. Analysis of Pollen Collected by Andrena flavipes Panzer (Hymenoptera: Andrenidae) in Sweet Cherry Orchards, Afyonkarahisar Province of Turkey. Psyche 2010, 2010, 160789. [Google Scholar] [CrossRef]
- Paxton, R.J.; Giovanetti, M.; Andrietti, F.; Scamoni, E.; Scanni, B. Mating in a Communal Bee, Andrena agilissima (Hymenoptera Andrenidae). Ethol. Ecol. Evol. 1999, 11, 371–382. [Google Scholar] [CrossRef]
- Polidori, C.; Rubichi, A.; Barbieri, V.; Trombino, L.; Donegana, M. Floral Resources and Nesting Requirements of the Ground-Nesting Social Bee, Lasioglossum malachurum (Hymenoptera: Halictidae), in a Mediterranean Semiagricultural Landscape. Psyche 2010, 2010, 851947. [Google Scholar] [CrossRef]
- Hajibabaei, M.; Janzen, D.H.; Burns, J.M.; Hallwachs, W.; Hebert, P.D.N. DNA Barcodes Distinguish Species of Tropical Lepidoptera. Proc. Natl. Acad. Sci. USA 2006, 103, 968–971. [Google Scholar] [CrossRef]
- Folmer, O.; Black, M.; Hoeh, W.; Lutz, R.; Vrijenhoek, R. DNA Primers for Amplification of Mitochondrial Cytochrome c Oxidase Subunit I from Diverse Metazoan Invertebrates. Mol. Mar. Biol. Biotechnol. 1994, 3, 294–299. [Google Scholar]
- Sequencher®, Version 5.4.6; DNA Sequence Analysis Software; Gene Codes Corporation: Ann Arbor, MI, USA, 2016.
- Larsson, A. AliView: A Fast and Lightweight Alignment Viewer and Editor for Large Datasets. Bioinformatics 2014, 30, 3276–3278. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An Ultra-Fast All-in-One FASTQ Preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Rochette, N.C.; Rivera-Colón, A.G.; Catchen, J.M. Stacks 2: Analytical Methods for Paired-end Sequencing Improve RADseq-based Population Genomics. Mol. Ecol. 2019, 28, 4737–4754. [Google Scholar] [CrossRef]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The Variant Call Format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- Lischer, H.E.L.; Excoffier, L. PGDSpider: An Automated Data Conversion Tool for Connecting Population Genetics and Genomics Programs. Bioinformatics 2012, 28, 298–299. [Google Scholar] [CrossRef]
- Cabreros, I.; Storey, J.D. A Likelihood-Free Estimator of Population Structure Bridging Admixture Models and Principal Components Analysis. Genetics 2019, 212, 1009–1029. [Google Scholar] [CrossRef]
- Pina-Martins, F.; Silva, D.N.; Fino, J.; Paulo, O.S. Structure_threader: An Improved Method for Automation and Parallelization of Programs Structure, FastStructure and MavericK on Multicore CPU Systems. Mol. Ecol. Resour. 2017, 17, e268–e274. [Google Scholar] [CrossRef]
- Jombart, T. Adegenet: A R Package for the Multivariate Analysis of Genetic Markers. Bioinformatics 2008, 24, 1403–1405. [Google Scholar] [CrossRef]
- R Core Team R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna, Austria. Available online: https://www.r-project.org (accessed on 26 March 2020).
- Malinsky, M.; Trucchi, E.; Lawson, D.J.; Falush, D. RADpainter and FineRADstructure: Population Inference from RADseq Data. Mol. Biol. Evol. 2018, 35, 1284–1290. [Google Scholar] [CrossRef] [PubMed]
- Goudet, J. HIERFSTAT, a Package for R to Compute and Test Hierarchical F-Statistics. Mol. Ecol. Notes 2005, 5, 184–186. [Google Scholar] [CrossRef]
- Kamvar, Z.N.; Tabima, J.F.; Grunwald, N.J. Poppr: An R Package for Genetic Analysis of Populations with Clonal, Partially Clonal, and/or Sexual Reproduction. PeerJ 2014, 2, e281. [Google Scholar] [CrossRef] [PubMed]
- Hedrick, P.W. A Standardized Genetic Differentiation Measure. Evolution 2005, 59, 1633–1638. [Google Scholar] [PubMed]
- Jost, L. GST and Its Relatives Do Not Measure Differentiation. Mol. Ecol. 2008, 17, 4015–4026. [Google Scholar] [CrossRef]
- Winter, D.J. Mmod: An R Library for the Calculation of Population Differentiation Statistics. Mol. Ecol. Resour. 2012, 12, 1158–1160. [Google Scholar] [CrossRef]
- Korunes, K.L.; Samuk, K. Pixy: Unbiased Estimation of Nucleotide Diversity and Divergence in the Presence of Missing Data. Mol. Ecol. Resour. 2021, 21, 1359–1368. [Google Scholar] [CrossRef]
- Exeler, N.; Kratochwil, A.; Hochkirch, A. Strong Genetic Exchange among Populations of a Specialist Bee, Andrena vaga (Hymenoptera: Andrenidae). Conserv. Genet. 2008, 9, 1233–1241. [Google Scholar] [CrossRef]
- Exeler, N.; Kratochwil, A.; Hochkirch, A. Does Recent Habitat Fragmentation Affect the Population Genetics of a Heathland Specialist, Andrena fuscipes (Hymenoptera: Andrenidae)? Conserv. Genet. 2010, 11, 1679–1687. [Google Scholar] [CrossRef]
- Jakobsson, M.; Edge, M.D.; Rosenberg, N.A. The Relationship Between FST and the Frequency of the Most Frequent Allele. Genetics 2013, 193, 515–528. [Google Scholar] [CrossRef]
- Meirmans, P.G.; Hedrick, P.W. Assessing Population Structure: FST and Related Measures. Mol. Ecol. Resour. 2011, 11, 5–18. [Google Scholar] [CrossRef]
- Friedel, A.; Paxton, R.J.; Soro, A. Spatial Patterns of Relatedness within Nesting Aggregations of the Primitively Eusocial Sweat Bee Lasioglossum malachurum. Insectes Soc. 2017, 64, 465–475. [Google Scholar] [CrossRef]
- Cruickshank, T.E.; Hahn, M.W. Reanalysis Suggests That Genomic Islands of Speciation Are Due to Reduced Diversity, Not Reduced Gene Flow. Mol. Ecol. 2014, 23, 3133–3157. [Google Scholar] [CrossRef]
- Zayed, A.; Packer, L. The Population Genetics of a Solitary Oligolectic Sweat Bee, Lasioglossum (Sphecodogastra) Oenotherae (Hymenoptera: Halictidae). Heredity 2007, 99, 397–405. [Google Scholar] [CrossRef]
- Su, T.; He, B.; Zhao, F.; Jiang, K.; Lin, G.; Huang, Z. Population Genomics and Phylogeography of Colletes Gigas, a Wild Bee Specialized on Winter Flowering Plants. Ecol. Evol. 2022, 12, e8863. [Google Scholar] [CrossRef]
- Smith, B.H.; Ayasse, M. Kin-Based Male Mating Preferences in Two Species of Halictine Bee. Behav. Ecol. Sociobiol. 1987, 20, 313–318. [Google Scholar] [CrossRef]
- Paxton, R.J.; Ayasse, M.; Field, J.; Soro, A. Complex Sociogenetic Organization and Reproductive Skew in a Primitively Eusocial Sweat Bee, Lasioglossum malachurum, as Revealed by Microsatellites. Mol. Ecol. 2002, 11, 2405–2416. [Google Scholar] [CrossRef]
- Paxton, R.J. Male Mating Behaviour and Mating Systems of Bees: An Overview. Apidologie 2005, 36, 145–156. [Google Scholar] [CrossRef]
- Paxton, R.J.; Thorén, P.A.; Tengö, J.; Estoup, A.; Pamilo, P. Mating Structure and Nestmate Relatedness in a Communal Bee, Andrena jacobi (Hymenoptera, Andrenidae), Using Microsatellites. Mol. Ecol. 1996, 5, 511–519. [Google Scholar] [CrossRef]
- Richards, M.H.; French, D.; Paxton, R.J. It’s Good to Be Queen: Classically Eusocial Colony Structure and Low Worker Fitness in an Obligately Social Sweat Bee. Mol. Ecol. 2005, 14, 4123–4133. [Google Scholar] [CrossRef]
- Soro, A.; Ayasse, M.; Zobel, M.U.; Paxton, R.J. Complex Sociogenetic Organization and the Origin of Unrelated Workers in a Eusocial Sweat Bee, Lasioglossum malachurum. Insectes Soc. 2009, 56, 55–63. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | N | Pop | Ho | He | FIS | Π |
|---|---|---|---|---|---|---|
| A. agilissima | 12 | 3 | 0.2104 ± 0.212 | 0.3173 | 0.3282 ± 0.533 | 0.2846 ± 0.235 |
| A. flavipes | 57 | 2 | 0.0807 ± 0.122 | 0.1187 | 0.2883 ± 0.429 | 0.0940 ± 0.151 |
| L. malachurum | 35 | 2 | 0.1806 ± 0.165 | 0.1978 | 0.1009 ± 0.316 | 0.1936 ± 0.186 |
| A. agilissima | |||
| West | Ebro | Baetic | |
| West | - | 0.3244 ± 0.202 | 0.3363 ± 0.180 |
| Ebro | 0.1114 | - | 0.3339 ± 0.200 |
| Baetic | 0.0926 | 0.0853 | - |
| A. flavipes | |||
| Iberian | Pyrenean | ||
| Iberian | - | 0.2362 ± 0.323 | |
| Pyrenean | 0.5611 | - | |
| L. malachurum | |||
| Ebro | Iberian | ||
| Ebro | - | 0.2088 ± 0.169 | |
| Iberian | 0.0663 | - | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sousa, P.d.; Henriques, A.; Silva, S.E.; Carvalheiro, L.G.; Smagghe, G.; Michez, D.; Wood, T.J.; Paulo, O.S. Genomic Patterns of Iberian Wild Bees Reveal Levels of Diversity, Differentiation and Population Structure, Supporting the “Refugia within Refugia” Hypothesis. Diversity 2023, 15, 746. https://doi.org/10.3390/d15060746
Sousa Pd, Henriques A, Silva SE, Carvalheiro LG, Smagghe G, Michez D, Wood TJ, Paulo OS. Genomic Patterns of Iberian Wild Bees Reveal Levels of Diversity, Differentiation and Population Structure, Supporting the “Refugia within Refugia” Hypothesis. Diversity. 2023; 15(6):746. https://doi.org/10.3390/d15060746
Chicago/Turabian StyleSousa, Paulo de, André Henriques, Sara E. Silva, Luísa G. Carvalheiro, Guy Smagghe, Denis Michez, Thomas J. Wood, and Octávio S. Paulo. 2023. "Genomic Patterns of Iberian Wild Bees Reveal Levels of Diversity, Differentiation and Population Structure, Supporting the “Refugia within Refugia” Hypothesis" Diversity 15, no. 6: 746. https://doi.org/10.3390/d15060746
APA StyleSousa, P. d., Henriques, A., Silva, S. E., Carvalheiro, L. G., Smagghe, G., Michez, D., Wood, T. J., & Paulo, O. S. (2023). Genomic Patterns of Iberian Wild Bees Reveal Levels of Diversity, Differentiation and Population Structure, Supporting the “Refugia within Refugia” Hypothesis. Diversity, 15(6), 746. https://doi.org/10.3390/d15060746