


Phylogenomic Analysis of Two Species of Parasenecio and Comparative Analysis within Tribe Senecioneae (Asteraceae)

Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Material, DNA Extraction, and Sequencing
2.2. CP Genome Assembly and Annotation
2.3. Repeat Sequence Analysis
2.4. Comparative Genome Analysis and Molecular Marker Identification
2.5. Phylogenetic Analysis
3. Results
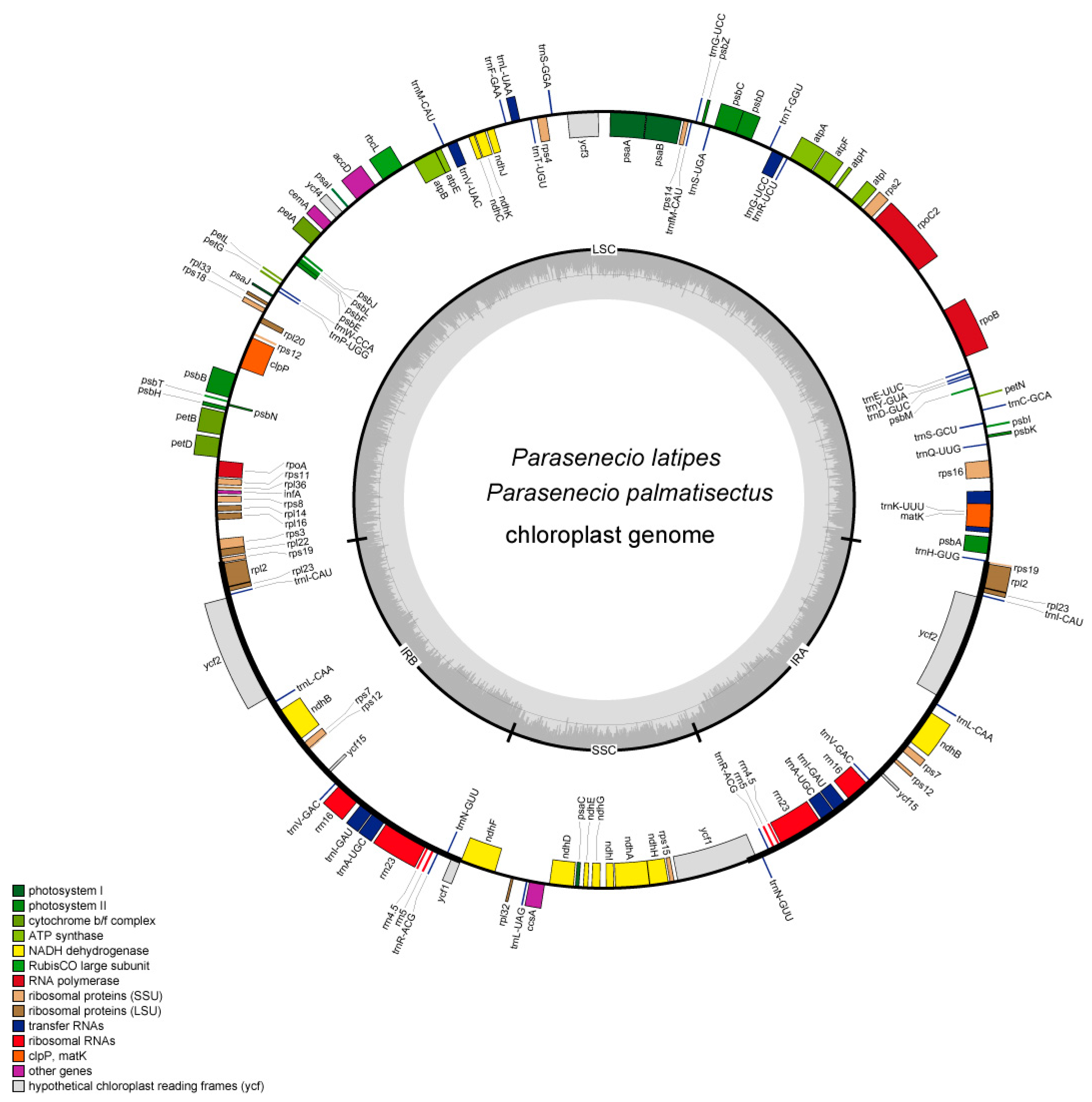
3.1. Features of the CP Genomes
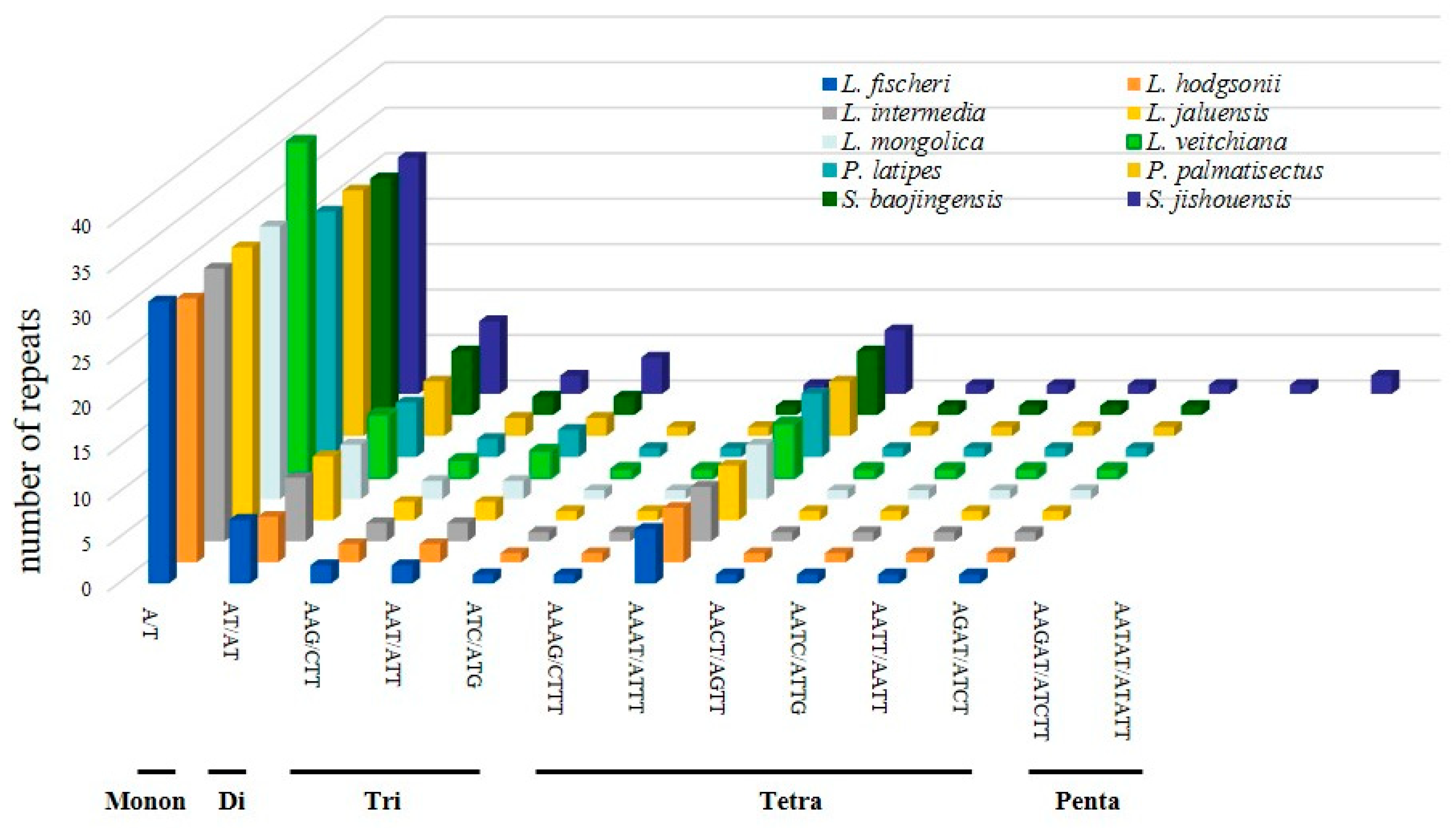
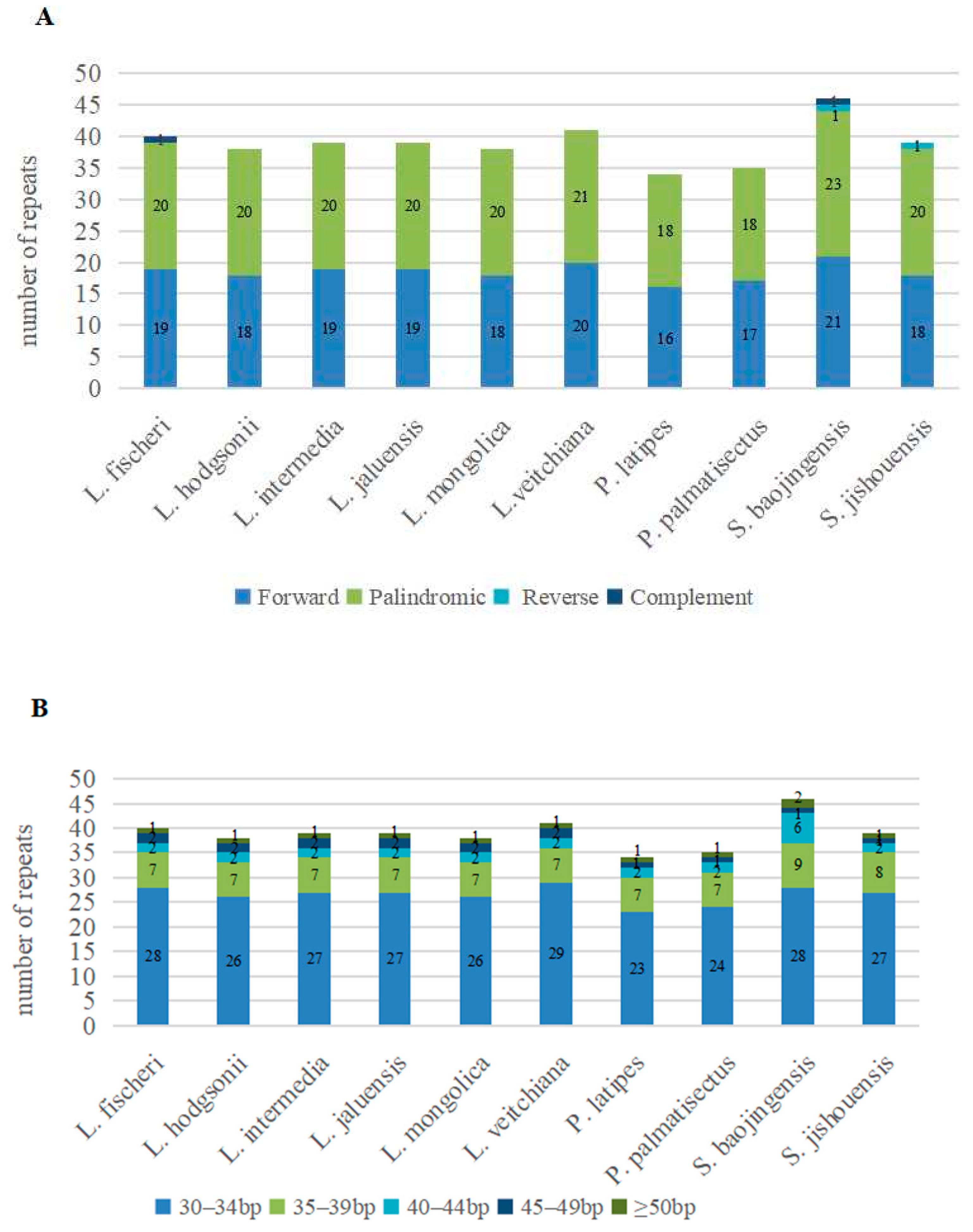
3.2. Repeat Sequences Analysis
3.3. IR Contraction and Expansion
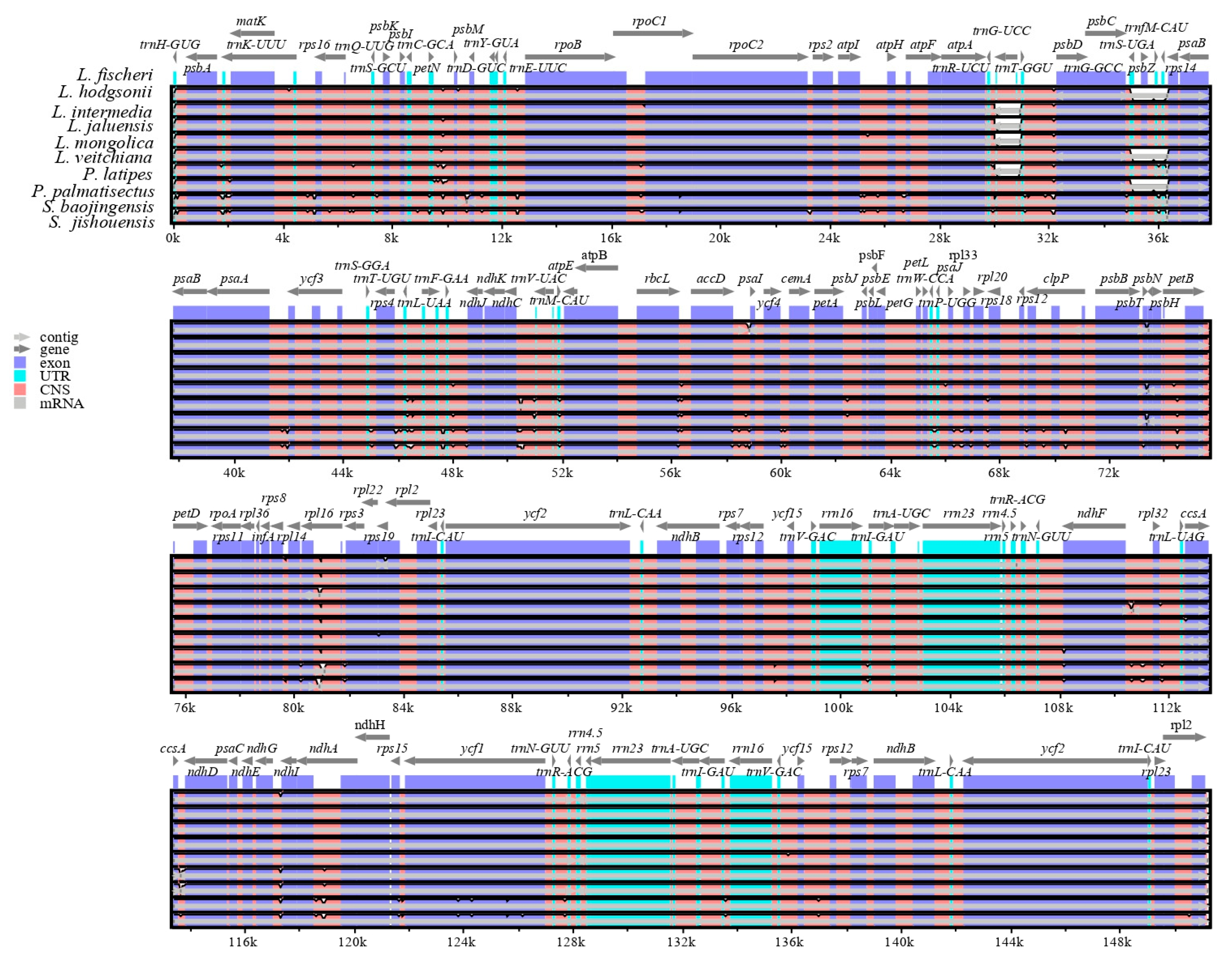
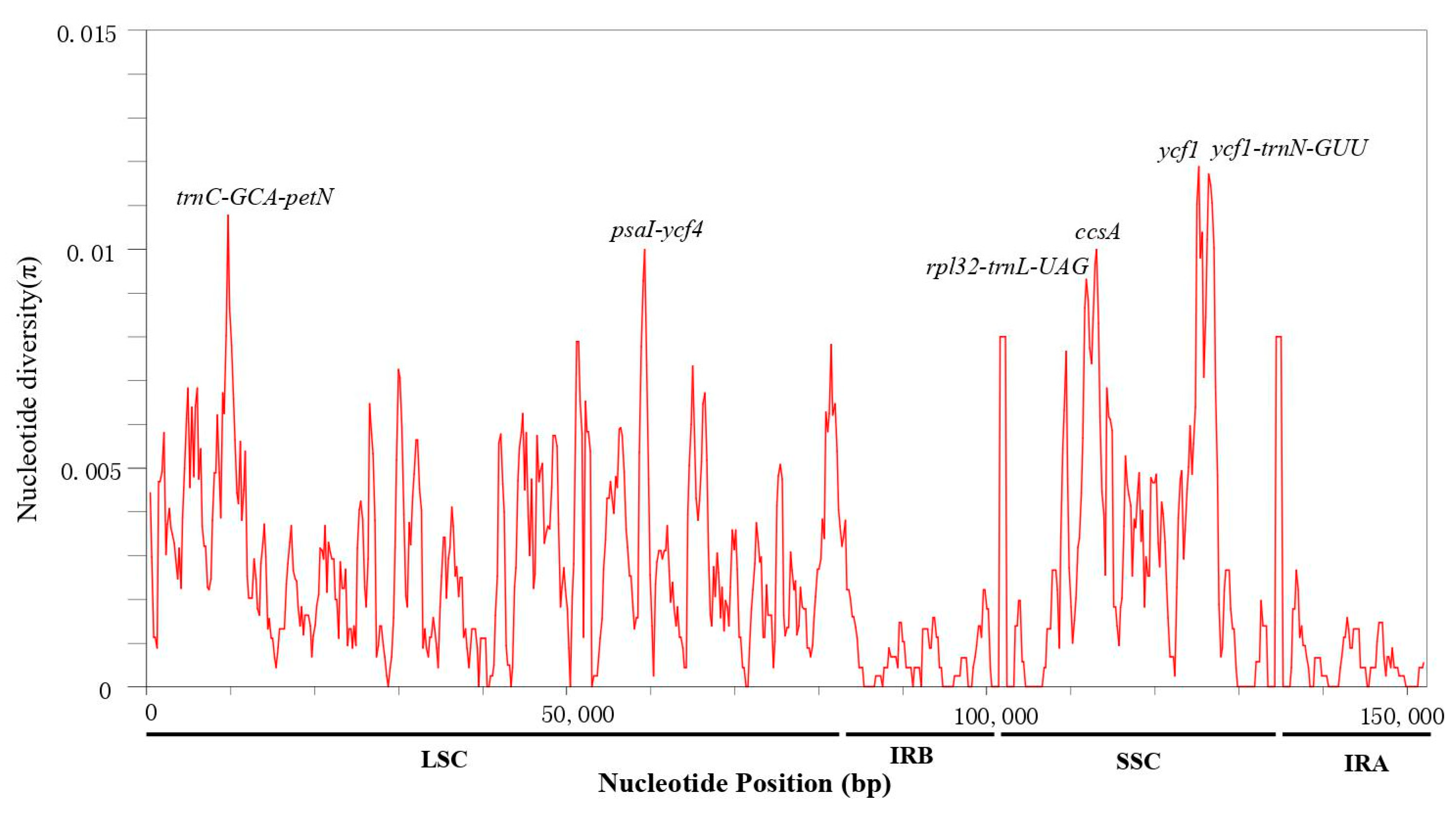
3.4. Sequence Divergence Analysis
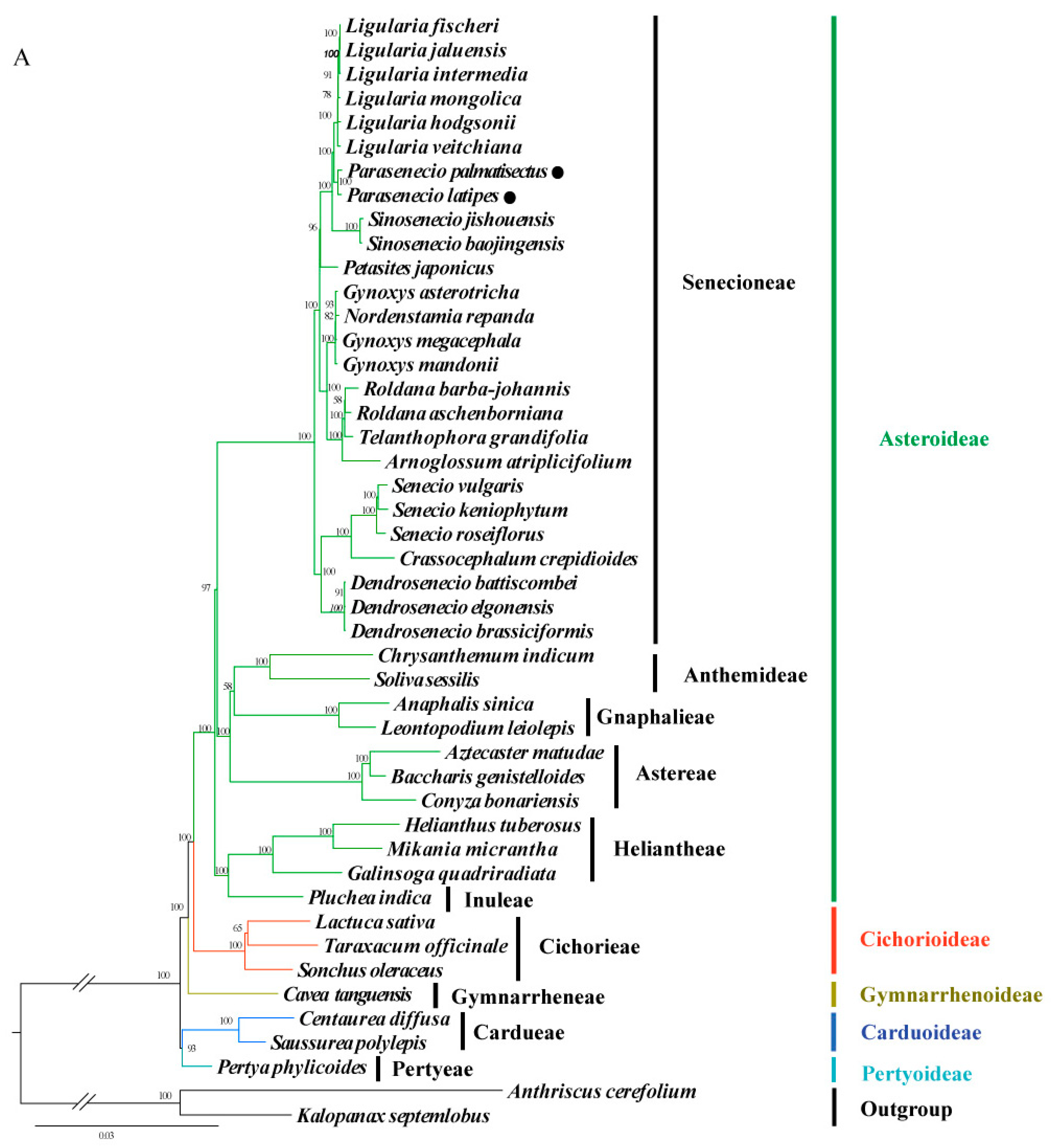
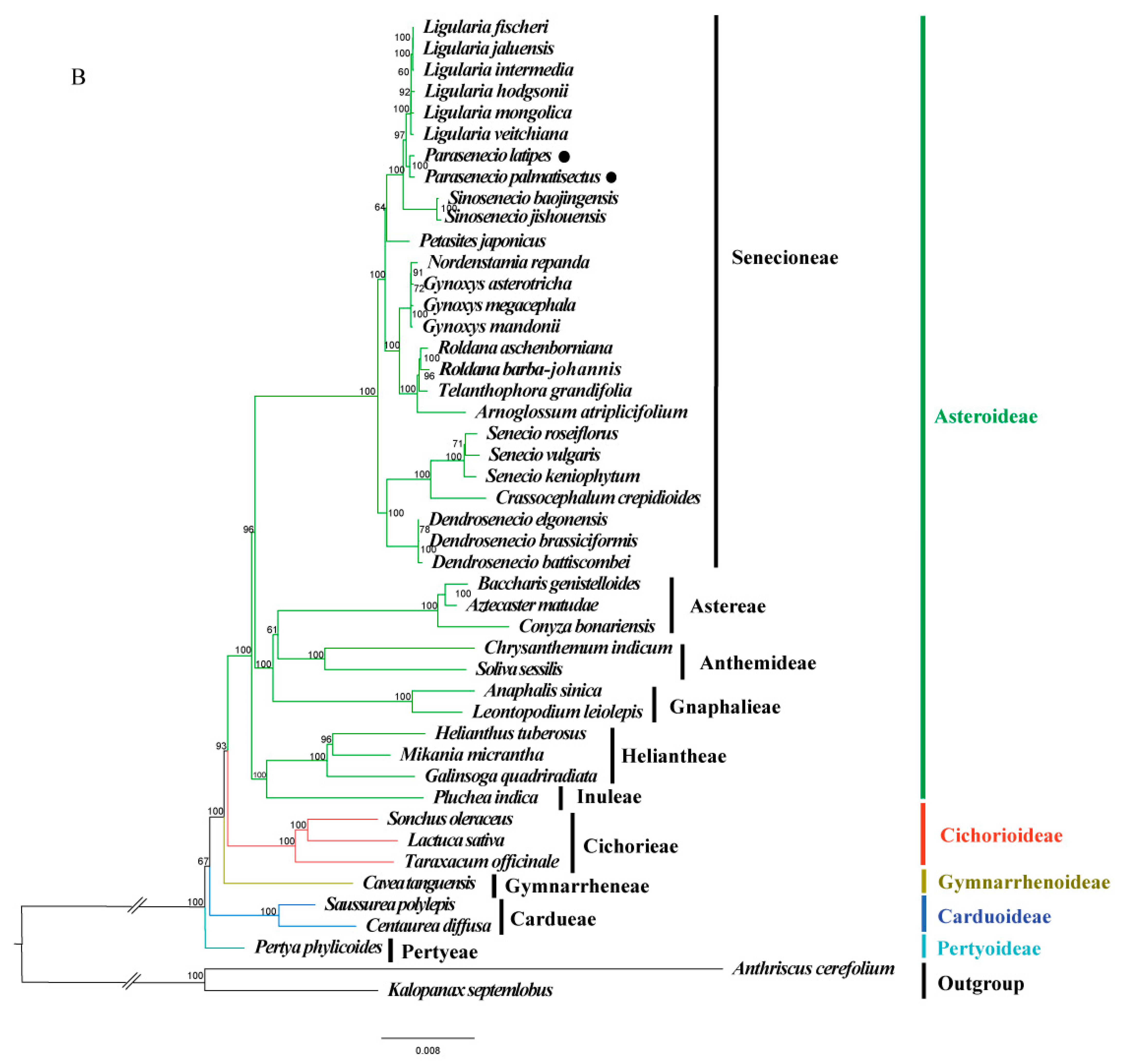
3.5. Phylogenomic Analysis
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- He, S.Z.; Peng, H. Parasenecio weiningensis (Asteraceae), a new species from Guizhou, SW China. Ann. Bot. Fenn. 2006, 43, 220–222. [Google Scholar]
- Chen, Y.L.; Liu, S.W.; Liu, Y.; Yang, Q.E.; Nordenstam, B.; Illarionova, I.D.; Jeffrey, C.; Koyama, H.; Vincent, L. Tribe Senecioneae. In Flora of China; Wu, Z.Y., Raven, P.H., Eds.; Science Press: Beijing, China; Missouri Botanical Garden Press: St. Louis, MO, USA, 2011. [Google Scholar]
- Chen, Y.S. A new species and a new combination in Parasenecio. Ann. Bot. Fenn. 2011, 48, 166–168. [Google Scholar] [CrossRef]
- Tang, M.; Ren, C.; Yang, Q.E. Parasenecio chenopodiifolius (Compositae-Senecioneae) is a Synotis and conspecific with S. otophylla based on evidence from morphology, cytology and ITS/ETS sequence data. Nord. J. Bot. 2014, 32, 824–835. [Google Scholar] [CrossRef]
- Xu, L.S.; Chen, Y.S. Parasenecio anhuiensis (Asteraceae, Senecioneae), a new species from eastern China. Phytotaxa 2016, 283, 188–194. [Google Scholar] [CrossRef]
- Ren, C.; Luo, J.P.; Hong, Y.; Yang, Q.E. Morphological and cytological data support the independent specific status of Parasenecio delphiniiphyllus (Asteraceae–Senecioneae) from China. Nord. J. Bot. 2015, 33, 455–464. [Google Scholar] [CrossRef]
- Liu, J.; Wang, Y.; Wang, A.; Hideaki, O.; Abbott, R. Radiation and diversification within the Ligularia-Cremanthodium-Parasenecio complex (Asteraceae) triggered by uplift of the Qinghai-Tibetan plateau. Mol. Phylogenet. Evol. 2006, 38, 31–49. [Google Scholar] [CrossRef]
- Pelser, P.B.; Nordenstam, B.; Kadereit, J.W.; Watson, L.E. An ITS phylogeny of Tribe Senecioneae (Asteraceae) and a new delimitation of Senecio L. Taxon 2007, 56, 1077–1104. [Google Scholar] [CrossRef]
- Wang, L.; Pelser, P.; Nordenstam, B.; Liu, J.Q. Strong incongruence between the ITS phylogeny and generic delimitation in the Nemosenecio-Sinosenecio-Tephroseris assemblage (Asteraceae: Senecioneae). Bot. Stud. 2009, 50, 435–442. [Google Scholar]
- Fu, Z.X.; Jiao, B.H.; Nie, B.; Zhang, G.J.; Gao, T.G. A comprehensive generic-level phylogeny of the sunflower family: Implications for the systematics of Chinese Asteraceae. J. Syst. Evol. 2016, 54, 416–437. [Google Scholar] [CrossRef]
- Ren, C.; Wang, L.; Nie, Z.L.; Johnson, G.; Yang, Q.E.; Wen, J. Development and phylogenetic utilities of a new set of single-/low-copy nuclear genes in Senecioneae (Asteraceae), with new insights into the tribal position and the relationships within subtribe Tussilagininae. Mol. Phylogenet. Evol. 2021, 162, 107202. [Google Scholar] [CrossRef]
- Ren, C.; Hong, Y.; Wang, L.; Yang, Q.E. Generic recircumscription of Parasenecio (Asteraceae: Senecioneae) based on nuclear ribosomal and plastid DNA sequences, with descriptions of two new genera. Bot. J. Linn. Soc. 2017, 184, 418–443. [Google Scholar] [CrossRef]
- Muhammad, I.; Wang, R.B.; Xiao, Y.Z.; Xie, Y.G.; Hassan, S.S.U.; Wu, G.J.; Qian, X.P.; Jin, H.Z. Chemical Constituents of Parasenecio quinquelobus. Chem. Nat. Compd. 2021, 57, 190–193. [Google Scholar] [CrossRef]
- Jin, A.; Duan, F.F.; Chang, J.L.; Liu, S.; Ruan, H.L. Chlorinated bisabolene sesquiterpenoids from the whole plant of Parasenecio rubescens. Fitoterapia 2022, 156, 105093. [Google Scholar] [CrossRef]
- Ayaz, A.; Huang, H.; Zheng, M.; Zaman, W.; Li, D.; Saqib, S.; Zhao, H.; Lü, S. Molecular cloning and functional analysis of GmLACS2-3 reveals its involvement in cutin and suberin biosynthesis along with abiotic stress tolerance. Int. J. Mol. Sci. 2021, 22, 9175. [Google Scholar] [CrossRef]
- Zaman, W.; Ye, J.; Ahmad, M.; Saqib, S.; Shinwari, Z.K.; Chen, Z. Phylogenetic exploration of traditional Chinese medicinal plants: A case study on Lamiaceae (angiosperms). Pak. J. Bot. 2022, 54, 1033–1040. [Google Scholar] [CrossRef]
- Shen, X.F.; Wu, M.L.; Liao, B.S.; Liu, Z.X.; Bai, R.; Xiao, S.M.; Li, X.W.; Zhang, B.L.; Xu, J.; Chen, S.L. Complete chloroplast genome sequence and phylogenetic analysis of the medicinal plant Artemisia annua. Molecules 2017, 22, 1330–1343. [Google Scholar] [CrossRef]
- Wen, F.; Wu, X.Z.; Li, T.J.; Jia, M.L.; Liu, X.S.; Liao, L. The complete chloroplast genome of Stauntonia chinensis and compared analysis revealed adaptive evolution of subfamily Lardizabaloideae species in China. BMC Genom. 2021, 22, 161. [Google Scholar] [CrossRef]
- Howe, C.J.; Barbrook, A.C.; Koumandou, V.L.; Nisbet, R.E.R.; Symington, H.A.; Wightman, T.F. Evolution of the chloroplast genome. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 2003, 358, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Shahzadi, I.; Mehmood, F.; Ali, Z.; Ahmed, I.; Mirza, B. Chloroplast genome sequences of Artemisia maritima and Artemisia absinthium: Comparative analyses, mutational hotspots in genus Artemisia and phylogeny in family Asteraceae. Genomics 2020, 112, 1454–1463. [Google Scholar] [CrossRef]
- Wicke, S.; Schneeweiss, G.M.; de Pamphilis, C.W.; Muller, K.F.; Quandt, D. The evolution of the plastid chromosome in land plants: Gene content, gene order, gene function. Plant Mol. Biol. 2011, 76, 273–297. [Google Scholar] [CrossRef] [PubMed]
- Gichira, A.W.; Avoga, S.; Li, Z.Z.; Hu, G.W.; Wang, Q.F.; Chen, J.M. Comparative genomics of 11 complete chloroplast genomes of Senecioneae (Asteraceae) species: DNA barcodes and phylogenetics. Bot. Stud. 2019, 60, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.X.; Liu, J.J.; Su, Y.; Li, M.L.; Xie, X.Y.; Su, J.J. Complete chloroplast genome sequence of Sonchus brachyotus helps to evolutionary relationships with related species of Asteraceae. BioMed Res. Int. 2021, 2021, 9410496. [Google Scholar] [CrossRef]
- Lan, Z.H.; Shi, Y.H.; Yin, Q.G.; Gao, R.R.; Liu, C.L.; Wang, W.T.; Tian, X.F.; Liu, J.W.; Nong, Y.Y.; Xiang, L.; et al. Comparative and phylogenetic analysis of complete chloroplast genomes from five Artemisia species. Front. Plant Sci. 2022, 13, 1049209. [Google Scholar] [CrossRef]
- Su, Y.J.; Huang, L.; Wang, Z.; Wang, T. Comparative chloroplast genomics between the invasive weed Mikania micrantha and its indigenous congener Mikania cordata: Structure variation, identification of highly divergent regions, divergence time estimation, and phylogenetic analysis. Mol. Phylogenet. Evol. 2018, 126, 181–195. [Google Scholar] [CrossRef]
- Zhang, X.; Deng, T.; Moore, M.J.; Ji, Y.H.; Lin, N.; Zhang, H.J.; Meng, A.P.; Wang, H.C.; Sun, Y.X.; Sun, H. Plastome phylogenomics of Saussurea (Asteraceae: Cardueae). BMC Plant Biol. 2019, 19, 290. [Google Scholar] [CrossRef]
- Chen, X.; Zhou, J.; Cui, Y.; Wang, Y.; Duan, B.; Yao, H. Identification of Ligularia herbs using the complete chloroplast genome as a super-barcode. Front. Pharmacol. 2018, 9, 695. [Google Scholar] [CrossRef]
- Zhang, Z.L.; Zhang, Y.; Song, M.F.; Guan, Y.H.; Ma, X.J. Species identification of Dracaena using the complete chloroplast genome as a super-barcode. Front. Pharmacol. 2019, 10, 1441. [Google Scholar] [CrossRef]
- Kim, K.-J.; Choi, K.-S.; Jansen, R.K. Two chloroplast DNA inversions originated simultaneously during the early evolution of the sunflower family (Asteraceae). Mol. Biol. Evol. 2005, 22, 1783–1792. [Google Scholar] [CrossRef]
- Walker, J.F.; Zanis, M.J.; Emery, N.C. Comparative analysis of complete chloroplast genome sequence and inversion variation in Lasthenia burkei (Madieae, Asteraceae). Am. J. Bot. 2014, 101, 722–729. [Google Scholar] [CrossRef]
- Choi, K.S.; Park, S. The complete chloroplast genome sequence of Aster spathulifolius (Asteraceae); genomic features and relationship with Asteraceae. Gene 2015, 572, 214–221. [Google Scholar] [CrossRef]
- Nagy, E.; Hegedûs, G.; Taller, J.; Kutasy, B.; Virág, E. Illumina sequencing of the chloroplast genome of common ragweed (Ambrosia artemisiifolia L.). Data Brief. 2017, 15, 606–611. [Google Scholar] [CrossRef]
- Tyagi, S.; Jung, J.-A.; Kim, J.S.; Won, S.Y. Comparative analysis of the complete chloroplast genome of mainland Aster spathulifolius and other Aster Species. Plants 2020, 9, 568. [Google Scholar] [CrossRef] [PubMed]
- Raman, G.; Park, K.T.; Kim, J.H.; Park, S. Characteristics of the completed chloroplast genome sequence of Xanthium spinosum: Comparative analyses, identification of mutational hotspots and phylogenetic implications. BMC Genom. 2020, 21, 855. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.H.; Yang, X.C.; Tian, X.Y.; Liu, X.F.; Lu, X.; Huang, C.P.; Fu, Z.X. The complete chloroplast genome sequence of the monotypic and enigmatic genus Cavea (tribe Gymnarrheneae) and a comparison with other species in Asteraceae. J. Genet. 2022, 101, 20. [Google Scholar] [CrossRef]
- Allen, G.; Flores-Vergara, M.; Krasynanski, S.; Kumar, S.; Thompson, W.F. A modified protocol for rapid DNA isolation from plant tissues using cetyltrimethylammonium bromide. Nat. Protoc. 2006, 1, 2320–2325. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Wick, R.R.; Schultz, M.B.; Zobel, J.; Holt, K.E. Bandage: Interactive visualisation of de novo genome assemblies. Bioinform. 2015, 31, 3350–3352. [Google Scholar] [CrossRef]
- Qu, X.J.; Moore, M.J.; Li, D.Z.; Yi, T.S. PGA: A software package for rapid, accurate, and flexible batch annotation of plastomes. Plant Methods 2019, 15, 50. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Greiner, S.; Lehwark, P.; Bock, R. Organellar Genome DRAW (OGDRAW) version 1.3.1: Expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 2019, 47, 59–64. [Google Scholar] [CrossRef]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. Reputer: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef]
- Beier, S.; Thiel, T.; Munch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinform. 2017, 33, 2583–2585. [Google Scholar] [CrossRef]
- Frazer, K.A.; Pachter, L.; Poliakov, A.; Rubin, E.M.; Dubchak, I. VISTA: Computational tools for comparative genomics. Nucleic Acids Res. 2004, 32, 273–279. [Google Scholar] [CrossRef]
- Amiryousefi, A.; Hyvönen, J.; Poczai, P. IRscope: An online program to visualize the junction sites of chloroplast genomes. Bioinform. 2018, 34, 3030–3031. [Google Scholar] [CrossRef]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 2019, 20, 1160–1166. [Google Scholar] [CrossRef]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Miller, M.A.; Pfeiffer, W.; Schwartz, T. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. In Proceedings of the Gateway Computing Environments Workshop (GCE), New Orleans, LA, USA, 14 November 2010; pp. 1–8. [Google Scholar]
- Chavhan, R.L.; Sable, S.; Narwade, A.V.; Hinge, V.R.; Kalbande, B.B.; Mukherjee, A.K.; Chakrabarty, P.K.; Kadam, U.S. Multiplex molecular marker-assisted analysis of significant pathogens of cotton (Gossypium Sp.). Biocatal. Agric. Biotechnol. 2023, 47, 102557. [Google Scholar] [CrossRef]
- Hinge, V.R.; Shaikh, I.M.; Chavhan, R.L.; Deshmukh, A.S.; Shelake, R.M.; Ghuge, S.A.; Dethe, A.M.; Suprasanna, P.; Kadam, U.S. Assessment of genetic diversity and volatile content of commercially grown banana (Musa spp.) cultivars. Sci. Rep. 2022, 12, 7979. [Google Scholar] [CrossRef]
- Upadhyay, A.; Kadam, U.S.; Chacko, P.; Karibasappa, G.S. Microsatellite and RAPD analysis of grape (Vitis spp.) accessions and identification of duplicates/misnomers in germplasm collection. Indian J. Hortic. 2010, 67, 8–15. [Google Scholar]
- Upadhyay, A.; Kadam, U.S.; Chacko, P.M.; Aher, L.; Karibasappa, G.S. Microsatellite analysis to differentiate clones of thompson seedless grapevine. Indian J. Hortic. 2010, 67, 260–263. [Google Scholar]
- Dong, W.P.; Xu, C.; Li, C.H.; Sun, J.H.; Zuo, Y.J.; Shi, S.; Cheng, T.; Guo, J.J.; Zhou, S.L. ycf1, the most promising plastid DNA barcode of land plants. Sci. Rep. 2015, 5, 8348. [Google Scholar] [CrossRef]
- Neubig, K.M.; Whitten, W.M.; Carlsward, B.S.; Blanco, M.A.; Endara, L.; Williams, N.H.; Moore, M. Phylogenetic utility of ycf1 in orchids: A plastid gene more variable than matK. Plant Syst. Evol. 2009, 277, 75–84. [Google Scholar] [CrossRef]
- Prince, L.M. Plastid primers for angiosperm phylogenetics and phylogeography. Appl. Plant Sci. 2015, 3, 1400085. [Google Scholar] [CrossRef] [PubMed]
- Salih, R.H.M.; Majeský, L.; Schwarzacher, T.; Gornall, R.; Heslop-Harrison, P. Complete chloroplast genomes from apomictic Taraxacum (Asteraceae): Identity and variation between three microspecies. PLoS ONE 2017, 12, e0168008. [Google Scholar] [CrossRef] [PubMed]
- Xie, D.F.; Tan, J.B.; Yu, Y.; Gui, L.J.; Su, D.M.; Zhou, S.D.; He, X.J. Insights into phylogeny, age and evolution of Allium (Amaryllidaceae) based on the whole plastome sequences. Ann. Bot. 2020, 125, 1039–1055. [Google Scholar] [CrossRef]
- Xie, D.F.; Yu, H.X.; Price, M.; Xie, C.; Deng, Y.Q.; Chen, J.P.; Yu, Y.; Zhou, S.D.; He, X.J. Phylogeny of Chinese Allium species in section Daghestanica and adaptive evolution of Allium (Amaryllidaceae, Allioideae) species revealed by the chloroplast complete genome. Front. Plant Sci. 2019, 10, 460. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | GenBank No. | Genome Size (bp) | LSC (bp) | IR (bp) | SSC (bp) | GC Content (%) | Number of Functional Genes | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| All | LSC | IR | SSC | Total | CDS | rRNAs | tRNAs | ||||||
| L. fischeri | MG729822 | 151,193 | 83,301 | 24,830 | 18,232 | 37.5 | 35.6 | 43.0 | 30.7 | 132 | 87 | 8 | 37 |
| L. hodgsonii | MF539929 | 151,136 | 83,253 | 24,833 | 18,217 | 37.5 | 35.6 | 43.0 | 30.7 | 132 | 87 | 8 | 37 |
| L. intermedia | MF539930 | 151,152 | 83,258 | 24,831 | 18,232 | 37.5 | 35.6 | 43.0 | 30.7 | 132 | 87 | 8 | 37 |
| L. jaluensis | MF539931 | 151,148 | 83,263 | 24,830 | 18,225 | 37.5 | 35.6 | 43.0 | 30.7 | 132 | 87 | 8 | 37 |
| L. mongolica | MF539932 | 151,118 | 83,244 | 24,830 | 18,214 | 37.5 | 35.6 | 43.0 | 30.7 | 132 | 87 | 8 | 37 |
| L. veitchiana | MF539933 | 151,253 | 83,330 | 24,838 | 18,247 | 37.5 | 35.6 | 43.0 | 30.7 | 132 | 87 | 8 | 37 |
| P. latipes | ON749759 | 151,185 | 83,308 | 24,828 | 18,221 | 37.5 | 35.6 | 43.0 | 30.8 | 131 | 86 | 8 | 37 |
| P. palmatisectus | ON749760 | 151,263 | 83,352 | 24,830 | 18,251 | 37.5 | 35.6 | 43.0 | 30.7 | 131 | 86 | 8 | 37 |
| S. baojingensis | MZ325394 | 151,315 | 83,445 | 24,849 | 18,172 | 37.4 | 35.5 | 43.0 | 30.6 | 132 | 87 | 8 | 37 |
| S. jishouensis | MT876597 | 151,257 | 83,373 | 24,853 | 18,178 | 37.4 | 35.5 | 43.0 | 30.6 | 134 | 89 | 8 | 37 |
| Category of Genes | Group of Genes | Name of Genes | Number of Genes |
|---|---|---|---|
| Photosynthesis | Subunits of photosystem I | psaA, psaB, psaC, psaI, psaJ | 5 |
| Subunits of photosystem II | psbA, psbB, psbC, psbD, psbE, psbF, psbH, psbI, psbJ, psbK, psbL, psbM, psbN, psbT, psbZ | 15 | |
| Subunits of NADH dehydrogenase | ndhA *, ndhB * (×2), ndhC, ndhD, ndhE, ndhF, ndhG, ndhH, ndhI, ndhJ, ndhK | 12 | |
| Subunits of cytochrome b/f complex | petA, petB *, petD *, petG, petL, petN | 6 | |
| Subunits of ATP synthase | atpA, atpB, atpE, atpF *, atpH, atpI | 6 | |
| Large subunit of rubisco | rbcL | 1 | |
| Self-replication | Proteins of large ribosomal subunit | rpl14, rpl16, rpl2 * (×2), rpl20, rpl22, rpl23(×2), rpl32, rpl33, rpl36 | 11 |
| Proteins of small ribosomal subunit | # rps19, rps11, rps12 ** (×2), rps14, rps15, rps16 *, rps18, rps19, rps2, rps3, rps4, rps7(×2), rps8 | 15 | |
| Subunits of RNA polymerase | rpoA, rpoB, rpoC2 | 3 | |
| Ribosomal RNAs | rrn16(×2), rrn23(×2), rrn4.5(×2), rrn5(×2) | 8 | |
| Transfer RNAs | trnA-UGC * (×2), trnC-GCA, trnD-GUC, trnE-UUC, trnF-GAA, trnG-UCC, trnG-UCC *, trnH-GUG, trnI-CAU(×2), trnI-GAU * (×2), trnK-UUU *, trnL-CAA(×2), trnL-UAA *, trnL-UAG, trnM-CAU, trnN-GUU(×2), trnP-UGG, trnQ-UUG, trnR-ACG(×2), trnR-UCU, trnS-GCU, trnS-GGA, trnS-UGA, trnT-GGU, trnT-UGU, trnV-GAC(×2), trnV-UAC *, trnW-CCA, trnY-GUA, trnfM-CAU | 37 | |
| Other genes | Maturase | matK | 1 |
| Protease | clpP ** | 1 | |
| Envelope membrane protein | cemA | 1 | |
| Acetyl-CoA carboxylase | accD | 1 | |
| c-type cytochrome synthesis gene | ccsA | 1 | |
| Translation initiation factor | infA | 1 | |
| Genes of unknown function | Conserved hypothetical chloroplast ORF | # ycf1, ycf1, ycf15(×2), ycf2(×2), ycf3 **, ycf4 | 8 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, X.; Luo, J.; Zhang, M.; Wang, Q.; Liu, J.; Wu, D.; Fu, Z. Phylogenomic Analysis of Two Species of Parasenecio and Comparative Analysis within Tribe Senecioneae (Asteraceae). Diversity 2023, 15, 563. https://doi.org/10.3390/d15040563
Liu X, Luo J, Zhang M, Wang Q, Liu J, Wu D, Fu Z. Phylogenomic Analysis of Two Species of Parasenecio and Comparative Analysis within Tribe Senecioneae (Asteraceae). Diversity. 2023; 15(4):563. https://doi.org/10.3390/d15040563
Chicago/Turabian StyleLiu, Xiaofeng, Junjia Luo, Mingke Zhang, Qiang Wang, Jie Liu, Die Wu, and Zhixi Fu. 2023. "Phylogenomic Analysis of Two Species of Parasenecio and Comparative Analysis within Tribe Senecioneae (Asteraceae)" Diversity 15, no. 4: 563. https://doi.org/10.3390/d15040563
APA StyleLiu, X., Luo, J., Zhang, M., Wang, Q., Liu, J., Wu, D., & Fu, Z. (2023). Phylogenomic Analysis of Two Species of Parasenecio and Comparative Analysis within Tribe Senecioneae (Asteraceae). Diversity, 15(4), 563. https://doi.org/10.3390/d15040563