

Multi-Locus Sequence Analysis Reveals the Genetic Diversity and Population Differentiation of Candidatus Liberibacter Asiaticus in China

Abstract
:1. Introduction
2. Materials and Methods
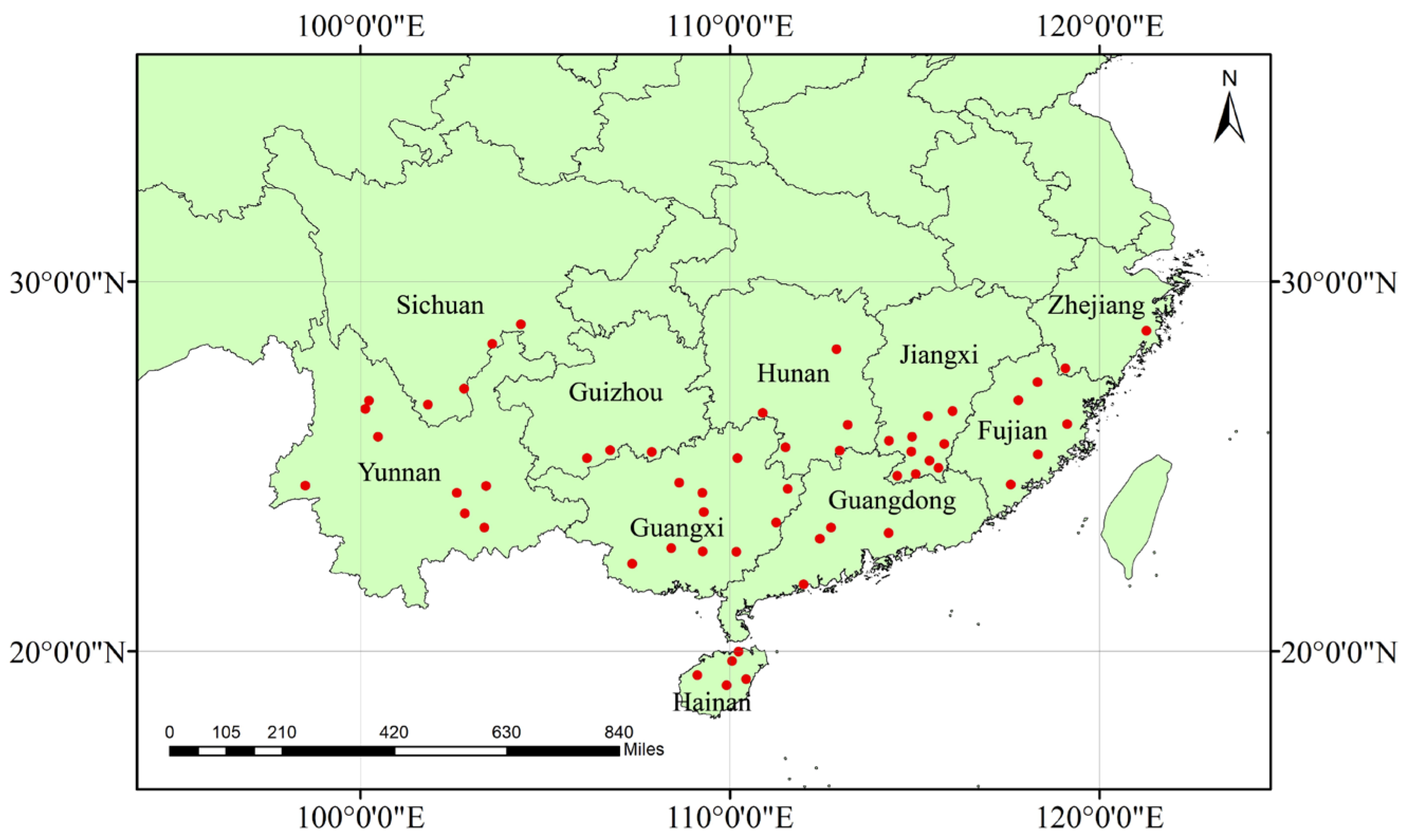
2.1. Sample Collection and DNA Extraction
2.2. Amplification and Purification of PCR Products
2.3. Sequence Assembly, Alignment, and Editing
2.4. Analysis of Genetic Diversity and Population Differentiation
2.5. Analysis of Demographic History
3. Results
3.1. Genetic Diversity and Population Differentiation
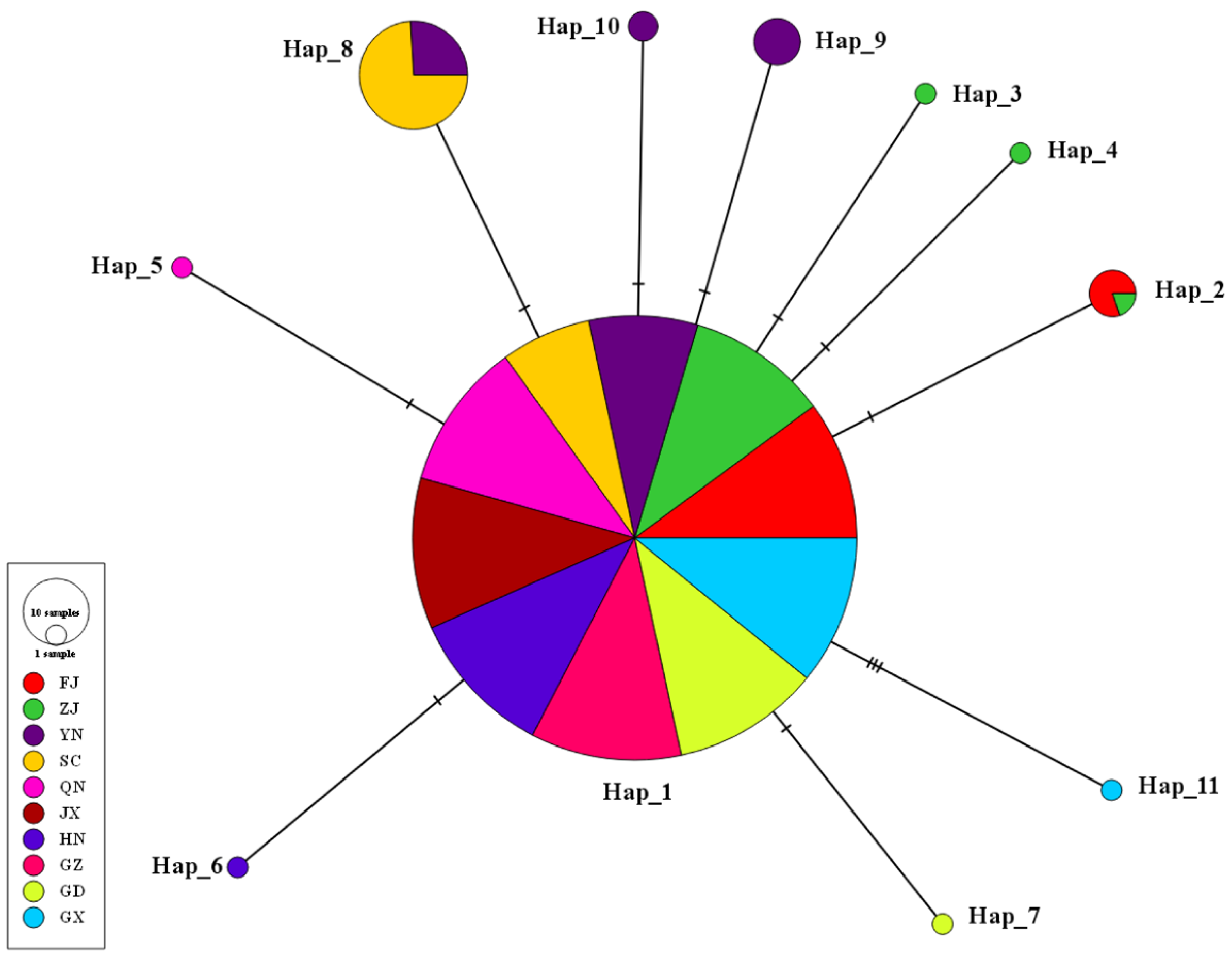
3.2. Identification and Distribution of Haplotypes
3.3. Gene Flow and Demographic History
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Bové, J.M.; Barros, A.P.D. Huanglongbing: A destructive, newly emerging, century-old disease of citrus. J. Plant Pathol. 2006, 88, 7–37. [Google Scholar]
- Morelli, M.; Takayassu, F.H.; Pacheco, C.D.; da Conceiçao, P.M.; Della Coletta, H.; de Azevedo, F.A. Impact of HLB on the physiological quality of citrus rootstock seeds and non-vertical ‘Candidatus Liberibacter asiaticus’ transmission. Trop. Plant Pathol. 2020, 45, 620–625. [Google Scholar] [CrossRef]
- Lu, J.M.; Delatte, H.; Reynaud, B.; Beattie, G.A.C.; Holford, P.; Cen, Y.J.; Wang, Y.J. Genome Sequence Resource of ‘Candidatus Liberibacter asiaticus’ from Diaphorina citri Kuwayama (Hemiptera: Liviidae) from La Reunion. Plant Dis. 2021, 105, 1171–1173. [Google Scholar] [CrossRef]
- Lin, H.; Pietersen, G.; Han, C.; Read, D.A.; Lou, B.; Gupta, G.; Civerolo, E.L. Complete Genome Sequence of “Candidatus Liberibacter africanus,” a Bacterium Associated with Citrus Huanglongbing. Genome Announc. 2015, 3, e00733-15. [Google Scholar] [CrossRef]
- Lin, H.; Coletta-Filho, H.D.; Han, C.S.; Lou, B.; Civerolo, E.L.; Machado, M.A.; Gupta, G. Draft Genome Sequence of “Candidatus Liberibacter americanus” Bacterium Associated with Citrus Huanglongbing in Brazil. Genome Announc. 2013, 1, e00275-13. [Google Scholar] [CrossRef]
- Yu, S.S.; Zhu, A.N.; Song, W.W.; Yan, W. Molecular Identification and Characterization of Two Groups of Phytoplasma and Candidatus Liberibacter Asiaticus in Single or Mixed Infection of Citrus maxima on Hainan Island of China. Biology 2022, 11, 869. [Google Scholar] [CrossRef]
- Blaustein, R.A.; Lorca, G.L.; Teplitski, M. Challenges for Managing Candidatus Liberibacter spp. (Huanglongbing Disease Pathogen): Current Control Measures and Future Directions. Phytopathology 2018, 108, 424–435. [Google Scholar] [CrossRef]
- Xie, Z.; Zhao, K.; Long, J.H.; Zhen, L.; Zou, X.P.; Chen, S.C. Comparative analysis of Wanjincheng orange leaf and root responses to ‘ Candidatus liberibacter asiaticus’ infection using leaf-disc grafting. Hortic. Plant J. 2021, 7, 401–410. [Google Scholar] [CrossRef]
- Hosseinzadeh, S.; Shams-Bakhsh, M.; Mann, M.; Fattah-Hosseini, S.; Bagheri, A.; Mehrabadi, M.; Heck, M. Distribution and Variation of Bacterial Endosymbiont and “Candidatus Liberibacter asiaticus” Titer in the Huanglongbing Insect Vector, Diaphorina citri Kuwayama. Microb. Ecol. 2019, 78, 206–222. [Google Scholar] [CrossRef]
- Pelz-Stelinski, K.S.; Brlansky, R.H.; Ebert, T.A.; Rogers, M.E. Transmission parameters for Candidatus liberibacter asiaticus by Asian citrus psyllid (Hemiptera: Psyllidae). J. Econ. Entomol. 2010, 103, 1531–1541. [Google Scholar] [CrossRef]
- Yang, C.Y.; Ancona, V. An Overview of the Mechanisms Against “Candidatus Liberibacter asiaticus”: Virulence Targets, Citrus Defenses, and Microbiome. Front. Microbiol. 2022, 13, 15. [Google Scholar] [CrossRef]
- Alquézar, B.; Carmona, L.; Bennici, S.; Peña, L. Engineering of citrus to obtain huanglongbing resistance. Curr. Opin. Biotechnol. 2021, 70, 196–203. [Google Scholar] [CrossRef]
- Alves, M.N.; Cifuentes-Arenas, J.C.; Raiol, L.L.; Ferro, J.A.; Peña, L. Early Population Dynamics of “Candidatus Liberibacter asiaticus” in Susceptible and Resistant Genotypes After Inoculation with Infected Diaphorina citri Feeding on Young Shoots. Front. Microbiol. 2021, 12, 683923. [Google Scholar] [CrossRef]
- Deng, X.L.; Gao, Y.D.; Chen, J.C.; Pu, X.L.; Kong, W.W.; Li, H.P. Curent Situation of “Candidatus Liberibacter asiaticus” in Guangdong, China, Where Citrus Huanglongbing Was First Described. J. Integr. Agric. 2012, 11, 424–429. [Google Scholar] [CrossRef]
- Hu, B.; Rao, M.J.; Deng, X.X.; Pandey, S.S.; Hendrich, C.; Ding, F.; Wang, N.; Xu, Q. Molecular signatures between citrus and Candidatus Liberibacter asiaticus. PLoS Pathog. 2021, 17, 22. [Google Scholar] [CrossRef]
- Zhou, C.Y. Status Citrus Huanglongbing China. Trop. Plant Pathol. 2020, 45, 279–284. [Google Scholar] [CrossRef]
- Cui, X.J.; Liu, K.H.; Huang, J.; Fu, S.M.; Chen, Q.D.; Liu, X.; Zhou, C.Y.; Wang, X.F. Population Diversity of ‘Candidatus Liberibacter asiaticus’ and Diaphorina citri in Sichuan: A Case Study for Huanglongbing Monitoring and Interception. Plant Dis. 2022, 106, 1632–1638. [Google Scholar] [CrossRef]
- Li, J.; Jin, Q.; Zhu, G.P.; Jiang, C.; Zhang, A.B. Phylogeography of Dendrolimus punctatus (Lepidoptera: Lasiocampidae): Population differentiation and last glacial maximum survival. Ecol. Evol. 2019, 9, 7480–7496. [Google Scholar] [CrossRef]
- Gao, F.L.; Chen, C.S.; Li, B.J.; Weng, Q.Y.; Chen, Q.H. The Gene Flow Direction of Geographically Distinct Phytophthora infestans Populations in China Corresponds with the Route of Seed Potato Exchange. Front. Microbiol. 2020, 11, 12. [Google Scholar] [CrossRef]
- Garrick, R.C.; Bonatelli, I.A.S.; Hyseni, C.; Morales, A.; Pelletier, T.A.; Perez, M.F.; Rice, E.; Satler, J.D.; Symula, R.E.; Thomé, M.T.C.; et al. The evolution of phylogeographic data sets. Mol. Ecol. 2015, 24, 1164–1171. [Google Scholar] [CrossRef]
- Card, D.C.; Schield, D.R.; Adams, R.H.; Corbin, A.B.; Perry, B.W.; Andrew, A.L.; Pasquesi, G.I.M.; Smith, E.N.; Jezkova, T.; Boback, S.M.; et al. Phylogeographic and population genetic analyses reveal multiple species of Boa and independent origins of insular dwarfism. Mol. Phylogenetics Evol. 2016, 102, 104–116. [Google Scholar] [CrossRef]
- Alam, M.A.; Li, H.X.; Hossain, A.; Li, M.J. Genetic Diversity of Wheat Stripe Rust Fungus Puccinia striiformis f. sp. tritici in Yunnan, China. Plants 2021, 10, 1735. [Google Scholar] [CrossRef]
- Zheng, Y.Q.; Huang, H.X.; Huang, Z.H.; Deng, X.L.; Zheng, Z.; Xu, M.R. Prophage region and short tandem repeats of “Candidatus Liberibacter asiaticus” reveal significant population structure in China. Plant Pathol. 2021, 70, 959–969. [Google Scholar] [CrossRef]
- Khanal, S.; Antony-Babu, S.; Gaire, S.P.; Zhou, X.G. Multi-Locus Sequence Analysis Reveals Diversity of the Rice Kernel Smut Populations in the United States. Front. Microbiol. 2022, 13, 12. [Google Scholar] [CrossRef]
- Das, A.K.; Chichghare, S.A.; Sharma, S.K.; Kumar, J.P.T.; Singh, S.; Baranwal, V.K.; Kumar, A.; Nerkar, S. Genetic diversity and population structure of ‘Candidatus Liberibacter asiaticus’ associated with citrus Huanglongbing in India based on the prophage types. World J. Microbiol. Biotechnol. 2021, 37, 14. [Google Scholar] [CrossRef]
- Zheng, Y.Q.; Zhang, J.X.; Li, Y.; Liu, Y.X.; Liang, J.Y.; Wang, C.; Fang, F.; Deng, X.L.; Zheng, Z. Pathogenicity and Transcriptomic Analyses of Two “Candidatus Liberibacter asiaticus” Strains Harboring Different Types of Phages. Microbiol. Spectr. 2023, 11, 16. [Google Scholar] [CrossRef]
- Gao, F.L.; Wu, B.; Zou, C.W.; Bao, Y.X.; Li, D.A.; Yao, W.; Powell, C.A.; Zhang, M.Q. Genetic Diversity of “Candidatus Liberibacter asiaticus” Based on Four Hypervariable Genomic Regions in China. Microbiol. Spectr. 2022, 10, 13. [Google Scholar] [CrossRef]
- Duan, Y.; Zhou, L.; Hall, D.G.; Li, W.; Doddapaneni, H.; Lin, H.; Liu, L.; Vahling, C.M.; Gabriel, D.W.; Williams, K.P.; et al. Complete genome sequence of citrus huanglongbing bacterium, ‘Candidatus Liberibacter asiaticus’ obtained through metagenomics. Mol. Plant Microbe Interact. 2009, 22, 1011–1020. [Google Scholar] [CrossRef]
- Hu, W.Z.; Wang, X.E.; Zhou, Y.; Li, Z.A.; Tang, K.Z.; Zhou, C.Y. Diversity of the omp gene in Candidatus Liberibacter asiaticus in China. J. Plant Pathol. 2011, 93, 211–214. [Google Scholar]
- Donnua, S.; Paradornuwat, A.; Chowpongpang, S.; Thaveechai, N. Genetic diversity of Candidatus Liberibacter asiaticus, the causal agent of citrus huanglongbing disease in Thailand using markers of dnaA, lpxD and zmpA genes. Thai J. Agric. Sci. 2012, 45, 171–180. [Google Scholar]
- Loto, F.; Coyle, J.F.; Padgett, K.A.; Pagliai, F.A.; Gardner, C.L.; Lorca, G.L.; Gonzalez, C.F. Functional characterization of LotP from Liberibacter asiaticus. Microb. Biotechnol. 2017, 10, 642–656. [Google Scholar] [CrossRef]
- Clark, K.J.; Pang, Z.Q.; Trinh, J.; Wang, N.; Ma, W.B. Sec-Delivered Effector 1 (SDE1) of ‘Candidatus Liberibacter asiaticus’ Promotes Citrus Huanglongbing. Mol. Plant-Microbe Interact. 2020, 33, 1394–1404. [Google Scholar] [CrossRef]
- Jagoueix, S.; Bové, J.M.; Garnier, M. PCR detection of the two ‘Candidatus’ liberobacter species associated with greening disease of citrus. Mol. Cell. Probes 1996, 10, 43–50. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1, vev003. [Google Scholar] [CrossRef]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA Sequence Polymorphism Analysis of Large Data Sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Excoffier, L.; Lischer, H.E.L. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar] [CrossRef]
- Pons, O.; Petit, R.J. Measuring and testing genetic differentiation with ordered versus unordered alleles. Genetics 1996, 144, 1237–1245. [Google Scholar] [CrossRef]
- Wright, S. Genetical structure of populations. Nature 1950, 166, 247–249. [Google Scholar] [CrossRef]
- Balloux, F.; Lugon-Moulin, N. The estimation of population differentiation with microsatellite markers. Mol. Ecol. 2002, 11, 155–165. [Google Scholar] [CrossRef]
- Leigh, J.W.; Bryant, D. POPART: Full-feature software for haplotype network construction. Methods Ecol. Evol. 2015, 6, 1110–1116. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Li, W.H. Unbiased estimation of the rates of synonymous and nonsynonymous substitution. J. Mol. Evol. 1993, 36, 96–99. [Google Scholar] [CrossRef]
- Tajima, F. DNA polymorphism in a subdivided population: The expected number of segregating sites in the two-subpopulation model. Genetics 1989, 123, 229–240. [Google Scholar] [CrossRef]
- Fu, Y.X. Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics 1997, 147, 915–925. [Google Scholar] [CrossRef]
- Fu, Y.X.; Li, W.H. Statistical tests of neutrality of mutations. Genetics 1993, 133, 693–709. [Google Scholar] [CrossRef]
- Yang, Z.; Zhang, L.; Zhao, J.F.; Zhang, X.K.; Wang, Y.; Li, T.S.; Zhang, W.; Zhou, Y. New geographic distribution and molecular diversity of Citrus chlorotic dwarf-associated virus in China. J. Integr. Agric. 2022, 21, 293–298. [Google Scholar] [CrossRef]
- Zheng, Z.; Chen, J.C.; Deng, X.L. Historical Perspectives, Management, and Current Research of Citrus HLB in Guangdong Province of China, Where the Disease has been Endemic for Over a Hundred Years. Phytopathology 2018, 108, 1224–1236. [Google Scholar] [CrossRef]
- Cai, L.; Jain, M.; Munoz-Bodnar, A.; Huguet-Tapia, J.C.; Gabriel, D.W. A synthetic ‘essentialome’ for axenic culturing of ‘Candidatus Liberibacter asiaticus’. BMC Res. Notes 2022, 15, 125. [Google Scholar] [CrossRef]
- Katoh, H.; Subandiyah, S.; Tomimura, K.; Okuda, M.; Su, H.J.; Iwanami, T. Differentiation of “Candidatus Liberibacter asiaticus” Isolates by Variable-Number Tandem-Repeat Analysis. Appl. Environ. Microbiol. 2011, 77, 1910–1917. [Google Scholar] [CrossRef]
- Wu, J.Y.; Zhou, D.Y.; Zhang, Y.; Mi, F.; Xu, J.P. Analyses of the Global Multilocus Genotypes of the Human Pathogenic Yeast Candida tropicalis. Front. Microbiol. 2019, 10, 14. [Google Scholar] [CrossRef]
- Tomimura, K.; Miyata, S.-I.; Furuya, N.; Kubota, K.; Okuda, M.; Subandiyah, S.; Hung, T.-H.; Su, H.-J.; Iwanami, T. Evaluation of genetic diversity among ‘Candidatus Liberibacter asiaticus’ isolates collected in Southeast Asia. Phytopathology 2009, 99, 1062–1069. [Google Scholar] [CrossRef]
- Jagoueix, S.; Bove, J.M.; Garnier, M. Comparison of the 16S/23S ribosomal intergenic regions of “Candidatus Liberobacter asiaticum” and “Candidatus Liberobacter africanum,” the two species associated with citrus huanglongbing (greening) disease. Int. J. Syst. Bacteriol. 1997, 47, 224–227. [Google Scholar] [CrossRef]
- Planet, P.; Jagoueix, S.; Bove, J.M.; Garnier, M. Detection and characterization of the African citrus greening liberobacter by amplification, cloning, and sequencing of the rplKAJL-rpoBC operon. Curr. Microbiol. 1995, 30, 137–141. [Google Scholar] [CrossRef]
- Liu, R.; Zhang, P.; Pu, X.L.; Xing, X.Q.; Chen, J.C.; Deng, X.L. Analysis of a Prophage Gene Frequency Revealed Population Variation of ‘Candidatus Liberibacter asiaticus’ from Two Citrus-Growing Provinces in China. Plant Dis. 2011, 95, 431–435. [Google Scholar] [CrossRef]
- Fu, S.; Bai, Z.; Su, H.; Liu, J.; Hartung, J.S.; Zhou, C.; Wang, X. Occurrence of prophage and historical perspectives associated with the dissemination of huanglongbing in mainland China. Plant Pathol. 2020, 69, 132–138. [Google Scholar] [CrossRef]
- Widyawan, A.; Ibrahim, Y.E.; Komy, M.H.E.; Dhafer, H.M.A.; Brown, J.K.; Al-Saleh, M.A. Differentiation of ‘Candidatus Liberibacter asiaticus’ in Saudi Arabia based on tandem repeat variability in genomic locus. J. King Saud Univ. Sci. 2023, 35, 8. [Google Scholar] [CrossRef]
- Ghosh, D.K.; Bhose, S.; Motghare, M.; Warghane, A.; Mukherjee, K.; Ghosh, D.K.; Sharma, A.K.; Ladaniya, M.S.; Gowda, S. Genetic Diversity of the Indian Populations of ‘Candidatus Liberibacter asiaticus’ Based on the Tandem Repeat Variability in a Genomic Locus. Phytopathology 2015, 105, 1043–1049. [Google Scholar] [CrossRef]
- Ma, W.; Liang, M.; Guan, L.; Xu, M.; Wen, X.; Deng, X.; Chen, J. Population Structures of ‘Candidatus Liberibacter asiaticus’ in Southern China. Phytopathology 2014, 104, 158–162. [Google Scholar] [CrossRef]
- Trubenová, B.; Hager, R. Social Selection and Indirect Genetic Effects in Structured populations. Evol. Biol. 2014, 41, 123–133. [Google Scholar] [CrossRef]
- Luebert, F.; Jacobs, P.; Hilger, H.H.; Muller, L.A.H. Evidence for nonallopatric speciation among closely related sympatric Heliotropium species in the Atacama Desert. Ecol. Evol. 2014, 4, 266–275. [Google Scholar] [CrossRef]
- Lenart, P.; Bienertova-Vasku, J.; Berec, L. Learning mitigates genetic drift. Sci. Rep. 2022, 12, 10. [Google Scholar] [CrossRef]
- Puttamuk, T.; Zhou, L.J.; Thaveechai, N.; Zhang, S.A.; Armstrong, C.M.; Duan, Y.P. Genetic Diversity of Candidatus Liberibacter asiaticus Based on Two Hypervariable Effector Genes in Thailand. PLoS ONE 2014, 9, 20. [Google Scholar] [CrossRef]
- de Paula, L.B.; Lin, H.; Stuchi, E.S.; Francisco, C.S.; Safady, N.G.; Coletta, H.D. Genetic diversity of ‘Candidatus Liberibacter asiaticus’ in Brazil analyzed in different geographic regions and citrus varieties. Eur. J. Plant Pathol. 2019, 154, 863–872. [Google Scholar] [CrossRef]
- Signor, S.A.; Abbasi, M.; Marjoram, P.; Nuzhdin, S.V. Social effects for locomotion vary between environments in Drosophila melanogaster females. Evolution 2017, 71, 1765–1775. [Google Scholar] [CrossRef]
- Chen, L.; Luo, J.Y.; Jin, M.L.; Yang, N.; Liu, X.G.; Peng, Y.; Li, W.Q.; Phillips, A.; Cameron, B.; Bernal, J.S.; et al. Genome sequencing reveals evidence of adaptive variation in the genus Zea. Nat. Genet. 2022, 54, 1736–1745. [Google Scholar] [CrossRef]
- Wu, F.N.; Jiang, H.Y.; Beattie, G.A.C.; Holford, P.; Chen, J.C.; Wallis, C.M.; Zheng, Z.; Deng, X.L.; Cen, Y.J. Population diversity of Diaphorina citri (Hemiptera: Liviidae) in China based on whole mitochondrial genome sequences. Pest Manag. Sci. 2018, 74, 2569–2577. [Google Scholar] [CrossRef]
- Zhou, L.J.; Powell, C.A.; Hoffman, M.T.; Li, W.B.; Fan, G.C.; Liu, B.; Lin, H.; Duan, Y.P. Diversity and Plasticity of the Intracellular Plant Pathogen and Insect Symbiont “Candidatus Liberibacter asiaticus” as Revealed by Hypervariable Prophage Genes with Intragenic Tandem Repeats. Appl. Environ. Microbiol. 2011, 77, 6663–6673. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Locus | Primers | Sequence (5′–3′) | Tm (°C) | Target Region | Reference |
|---|---|---|---|---|---|
| CLIBASIA_02570 | F | AACAGCCTCTTCACCTTCGA | 55 | DnaA | This study |
| R | TGGCGAGGTCTTACAACAGT | ||||
| CLIBASIA_05315 | F | AGGAAATATCGTGCGTAAAA | 45 | SDE1 | [32] |
| R | GCTCCAACATTTTTCTATGG | ||||
| CLIBASIA_03315 | F | GAGGTGCCACGAATGTAGGT | 55 | OMP | This study |
| R | TGGCAATTGCGAAATATTGA | ||||
| CLIBASIA_03720 | F | CCTCGCGTTACTAAGGATGG | 56 | GroEL | This study |
| R | CCTCGCGTTACTAAGGATGG |
| POP | S | Pi | K | H | Hd |
|---|---|---|---|---|---|
| Total | 12 | 0.00005 ± 0.00001 | 0.182 | 11 | 0.169 ± 0.022 |
| Fujian (FJ) | 1 | 0.00004 ± 0.00002 | 0.15 | 2 | 0.150 ± 0.065 |
| Guangdong (GD) | 1 | 0.00001 ± 0.00001 | 0.04 | 2 | 0.040 ± 0.038 |
| Guangxi (GX) | 4 | 0.00002 ± 0.00022 | 0.08 | 3 | 0.040 ± 0.027 |
| Guizhou (GZ) | 0 | 0 | 0 | 1 | 0 |
| Hunan (HN) | 1 | 0.00001 ± 0.00001 | 0.04 | 2 | 0.040 ± 0.038 |
| Jiangxi (JX) | 0 | 0 | 0 | 1 | 0 |
| Hainan (QN) | 1 | 0.00001 ± 0.00001 | 0.04 | 2 | 0.040 ± 0.038 |
| Sichuan (SC) | 1 | 0.00014 ± 0.00001 | 0.49 | 2 | 0.490 ± 0.031 |
| Yunnan (YN) | 3 | 0.00015 ± 0.00003 | 0.508 | 4 | 0.460 ± 0.078 |
| Zhejiang (ZJ) | 3 | 0.00003 ± 0.00019 | 0.12 | 4 | 0.118 ± 0.062 |
| GENES | S | Pi | K | H | Hd | dN | dS |
|---|---|---|---|---|---|---|---|
| omp | 5 | 0.00004 ± 0.00002 | 0.036 | 4 | 0.028 ± 0.010 | 1.80 × 10−6 | 1.40 × 10−5 |
| DnaA | 0 | 0 | 0 | 1 | 0 | 0 | 0 |
| GroEL | 2 | 0.000000633 ± 0.00000 | 0.008 | 3 | 0.008 ± 0.00003 | 4.31 × 10−6 | 1.40 × 10−5 |
| SDE1 | 5 | 0.00031 ± 0.00005 | 0.138 | 6 | 0.136 ± 0.00042 | 0 | 4.38 × 10−4 |
| Locus | Source of Variation | d.f. | Sum of Squares | Variance Components | Percentage of Variation % | FST |
|---|---|---|---|---|---|---|
| omp | Among populations | 9 | 0.540 | 0.00082 Va | 4.08 | 0.04082 |
| Within populations | 490 | 9.400 | 0.01918 Vb | 95.92 | ||
| Total | 499 | 9.940 | 0.02000 | 100.00 | ||
| DnaA | Among populations | 9 | 0.000 | 0.00000 Va | 0.00 | 0.00000 |
| Within populations | 490 | 0.000 | 0.0000 Vb | 0.00 | ||
| Total | 499 | 0.000 | 0.0000 | 0.00 | ||
| GroEL | Among populations | 9 | 0.036 | 0.00000 Va | 0.00 | 0.00000 |
| Within populations | 490 | 1.960 | 0.00400 Vb | 100.00 | ||
| Total | 499 | 1.996 | 0.00400 | 100.00 | ||
| SDE1 | Among populations | 9 | 7.920 | 0.01652 Va | 23.35 | 0.23354 |
| Within populations | 490 | 26.560 | 0.05420 Vb | 76.65 | ||
| Total | 499 | 34.480 | 0.07072 | 100.00 | ||
| Multi-locus | Among populations | 9 | 8.496 | 0.01733 Va | 18.30 | 0.18296 |
| Within populations | 490 | 37.920 | 0.07739 Vb | 81.70 | ||
| Total | 499 | 46.416 | 0.09472 | 100.00 |
| Population | N | Neutrality Test | |
|---|---|---|---|
| Tajima’s D | Fu’s FS | ||
| Total | 500 | −1.98492 * | −15.17394 * |
| FJ | 50 | −0.4396 | −0.07635 |
| ZJ | 50 | −1.69603 * | −4.30079 * |
| YN | 50 | −0.49979 | −0.79565 |
| SC | 50 | 1.60401 | 1.89461 |
| QN | 50 | −1.1028 | −1.63631 * |
| JX | 50 | NA | NA |
| HN | 50 | −1.1028 | −1.63631 * |
| GZ | 50 | NA | NA |
| GD | 50 | −1.1028 | −1.63631 * |
| GX | 50 | −1.69603 * | 0.00841 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
You, P.; Bao, M.; Amir, M.B.; Zhou, J.; Yang, J.; Fang, S.; Yi, L. Multi-Locus Sequence Analysis Reveals the Genetic Diversity and Population Differentiation of Candidatus Liberibacter Asiaticus in China. Diversity 2023, 15, 1161. https://doi.org/10.3390/d15121161
You P, Bao M, Amir MB, Zhou J, Yang J, Fang S, Yi L. Multi-Locus Sequence Analysis Reveals the Genetic Diversity and Population Differentiation of Candidatus Liberibacter Asiaticus in China. Diversity. 2023; 15(12):1161. https://doi.org/10.3390/d15121161
Chicago/Turabian StyleYou, Ping, Minli Bao, Muhammad Bilal Amir, Jun Zhou, Jin Yang, Shujie Fang, and Long Yi. 2023. "Multi-Locus Sequence Analysis Reveals the Genetic Diversity and Population Differentiation of Candidatus Liberibacter Asiaticus in China" Diversity 15, no. 12: 1161. https://doi.org/10.3390/d15121161
APA StyleYou, P., Bao, M., Amir, M. B., Zhou, J., Yang, J., Fang, S., & Yi, L. (2023). Multi-Locus Sequence Analysis Reveals the Genetic Diversity and Population Differentiation of Candidatus Liberibacter Asiaticus in China. Diversity, 15(12), 1161. https://doi.org/10.3390/d15121161