
Spatial Variations of Aquatic Bacterial Community Structure and Co-Occurrence Patterns in a Coal Mining Subsidence Lake

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
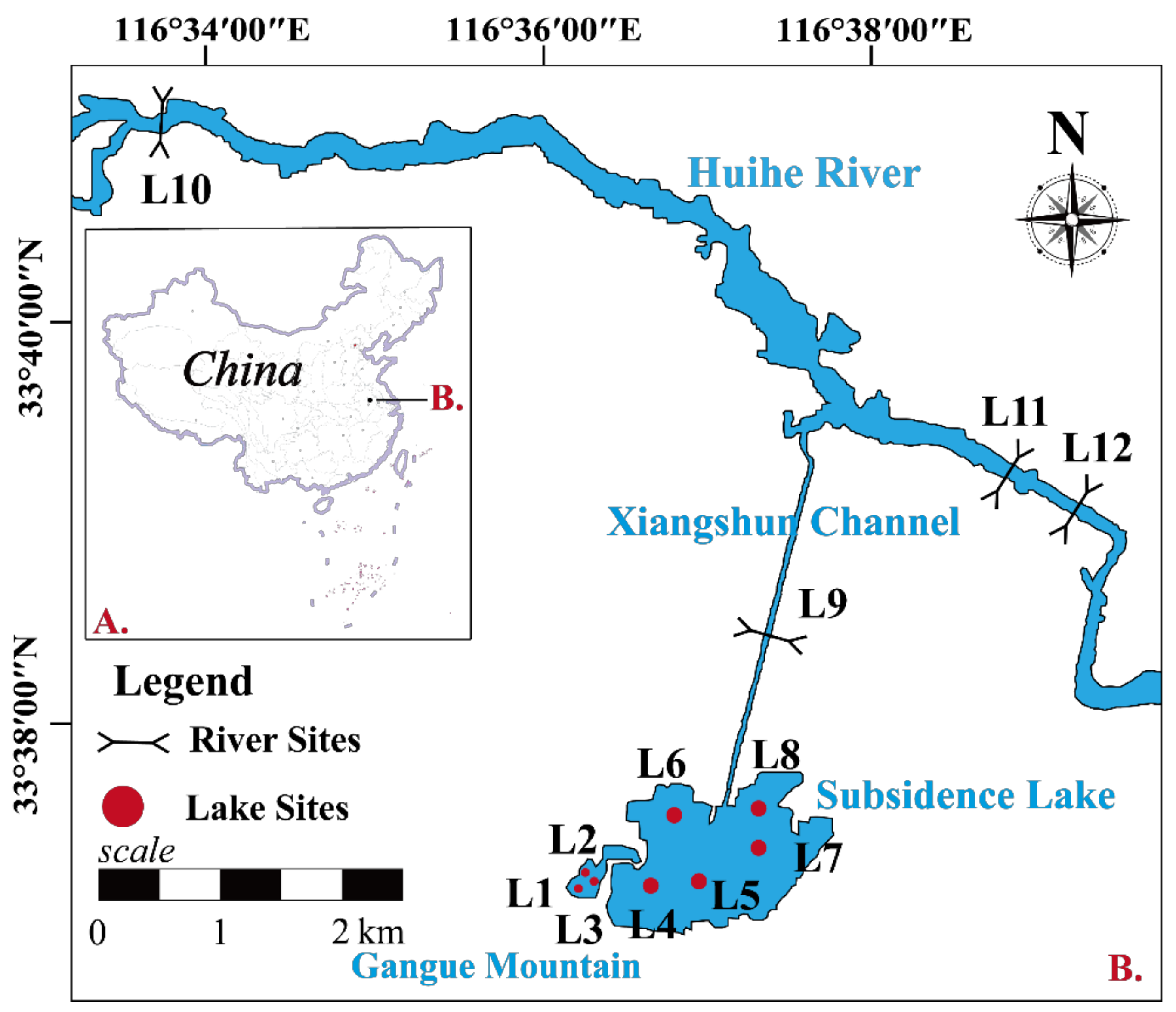
2.1. Site Description and Sample Collection
2.2. Analysis of Physicochemical Indices
2.3. DNA Extraction and PCR Amplification
2.4. Sequence Analyses
2.5. Statistical Analyses
3. Results
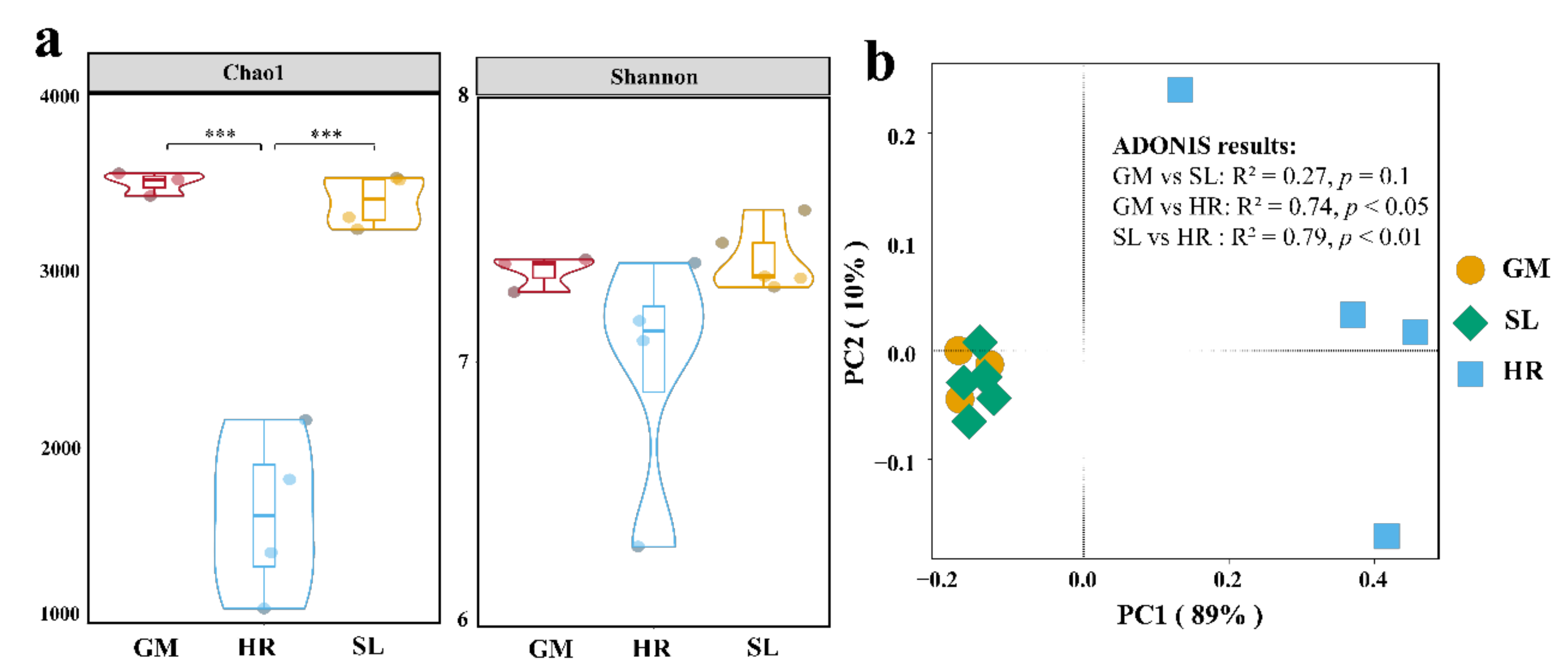
3.1. Alpha and Beta Diversity of the Bacterial Community
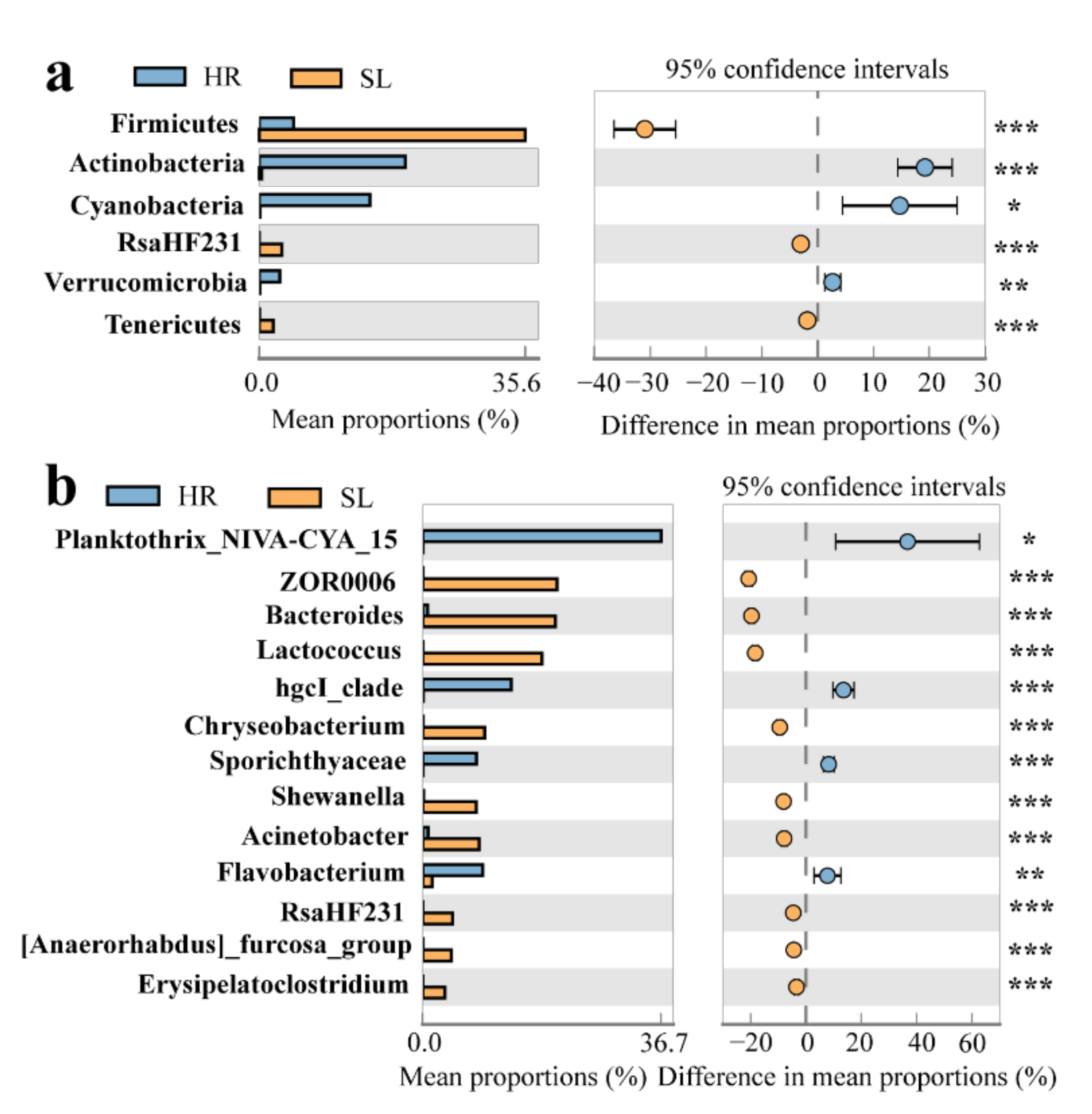
3.2. Taxonomic Composition of the Bacterial Community
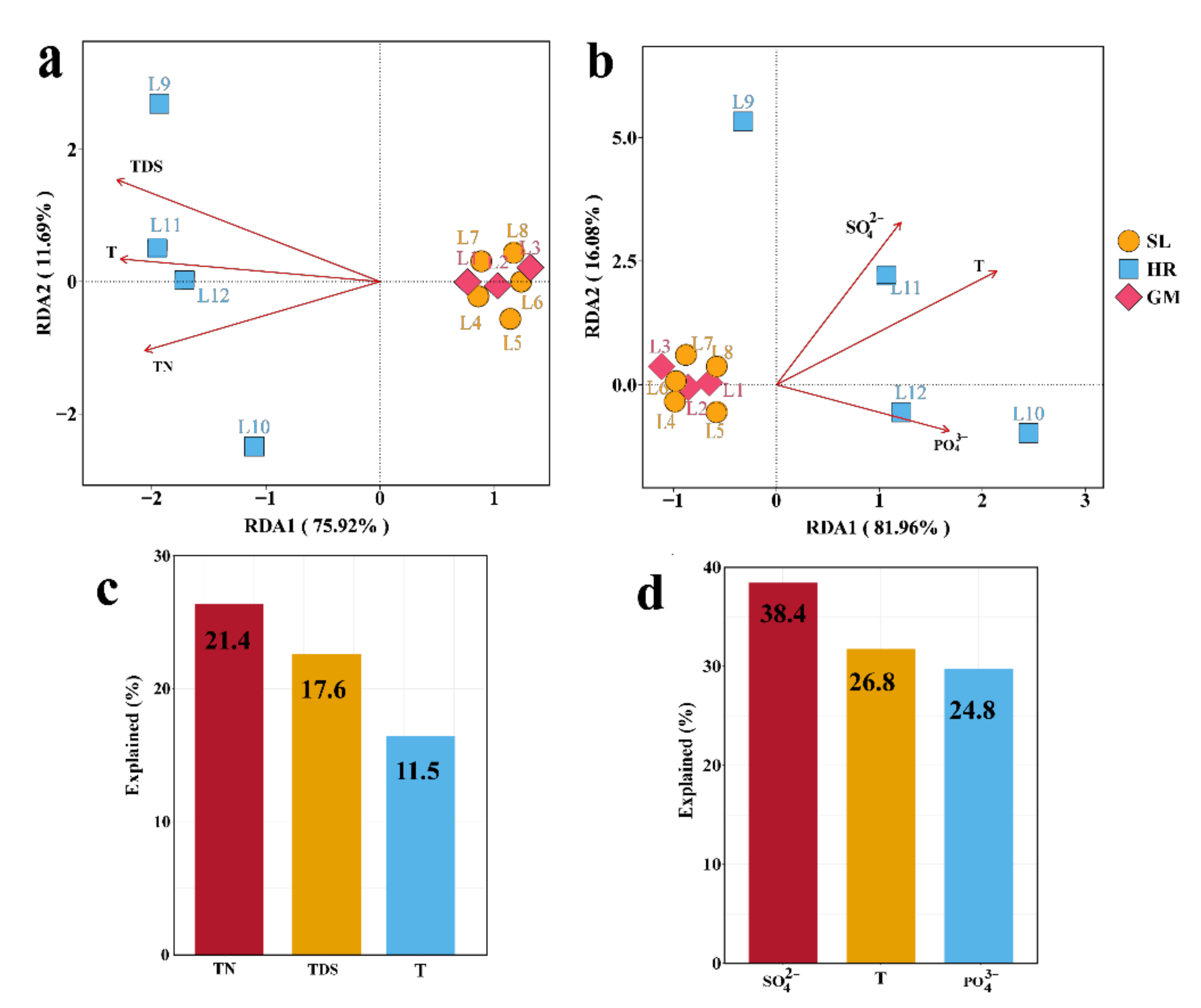
3.3. Physiochemical Properties and Redundancy Analysis
3.4. Distribution Characteristics of Sulfur Metabolism Genes in Different Samples
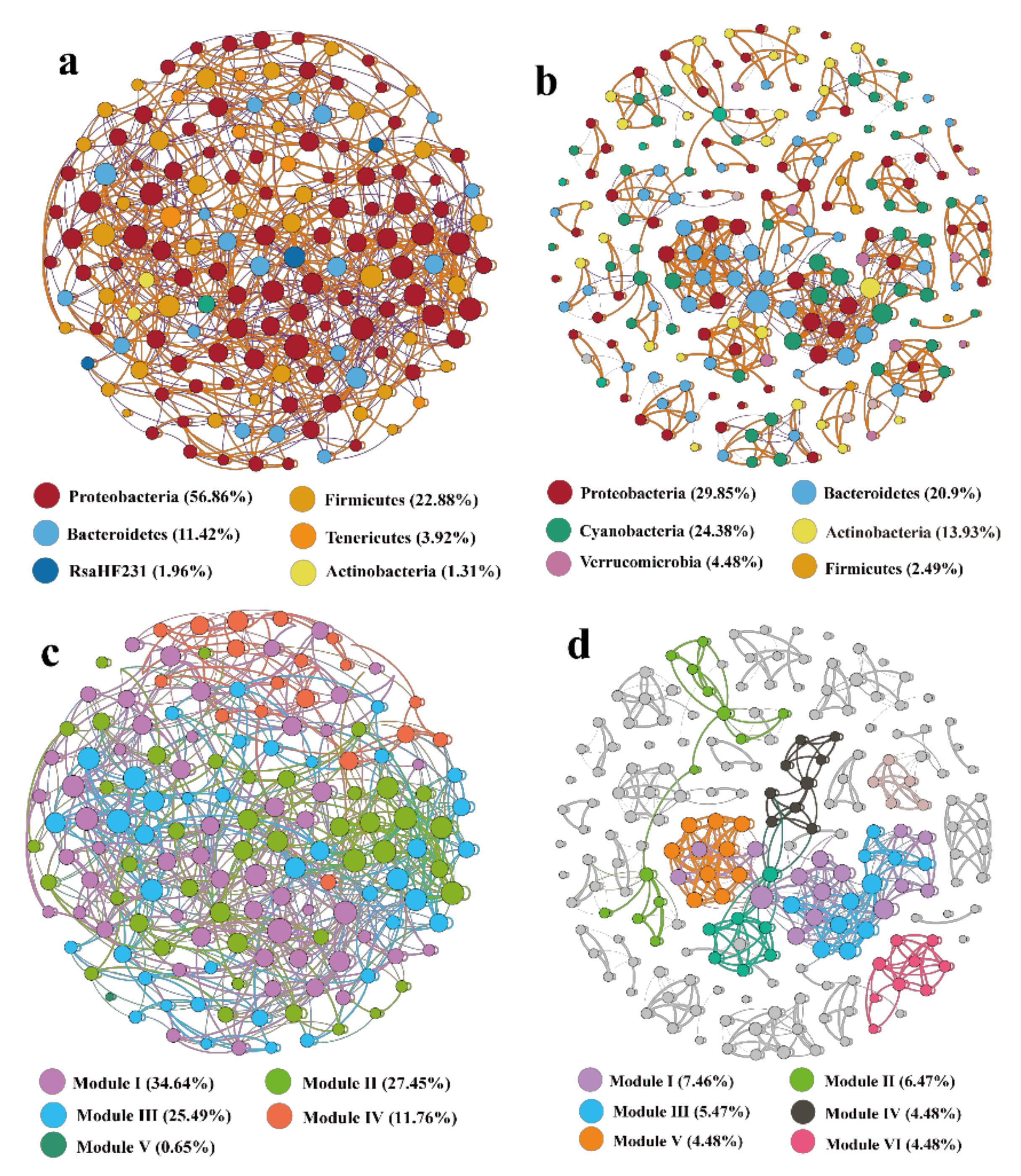
3.5. Bacterial Community Co-Occurrence Network Analysis
4. Discussion
4.1. Variation of Bacterial Diversity in Subsidence Lake and Its Connected River
4.2. Variations of Bacterial Community Composition at Different Sites
4.3. Predicted Functional Potential of Bacterial Communities Associated with Sulfur Metabolism
4.4. Differences in Microbiome Complexity and Keystone Taxa in the Subsidence Lake and Its Connected River
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liu, Y.; Zhu, J.; Ye, C.; Zhu, P.; Ba, Q.; Pang, J.; Shu, L. Effects of biochar application on the abundance and community composition of denitrifying bacteria in a reclaimed soil from coal mining subsidence area. Sci. Total Environ. 2018, 625, 1218–1224. [Google Scholar] [CrossRef]
- Sun, S.; Sun, H.; Zhang, D.; Zhang, J.; Cai, Z.; Qin, G.; Song, Y. Response of Soil Microbes to Vegetation Restoration in Coal Mining Subsidence Areas at Huaibei Coal Mine, China. Int. J. Environ. Res. Public Health 2019, 16, 1757. [Google Scholar] [CrossRef]
- HU, Z.-Q.; WANG, X.-J.; HE, A.-M. Distribution Characteristic and Development Rules of Ground Fissures Due to Coal Mining in Windy and Sandy Region. J. China Coal Soc. 2014, 39, 11–18. [Google Scholar]
- Zhou, D.-W.; Wu, K.; Cheng, G.-L.; Li, L. Mechanism of mining subsidence in coal mining area with thick alluvium soil in China. Arab. J. Geosci. 2014, 8, 1855–1867. [Google Scholar] [CrossRef]
- Pei, W.; Yao, S.; Knight, J.F.; Dong, S.; Pelletier, K.; Rampi, L.P.; Wang, Y.; Klassen, J. Mapping and detection of land use change in a coal mining area using object-based image analysis. Environ. Earth Sci. 2017, 76, 125. [Google Scholar] [CrossRef]
- Mason, T.J.; Krogh, M.; Popovic, G.C.; Glamore, W.; Keith, D.A. Persistent effects of underground longwall coal mining on freshwater wetland hydrology. Sci. Total Environ. 2021, 772, 144772. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; Liu, G.; Zheng, L.; Chou, C.-L. Characteristics of coal quality and their relationship with coal-forming environment: A case study from the Zhuji exploration area, Huainan coalfield, Anhui, China. Energy 2010, 35, 423–435. [Google Scholar] [CrossRef]
- Zheng, L.; Liu, X.; Tang, Q.; Ou, J. Lead Pollution and Isotope Tracing of Surface Sediments in the Huainan Panji Coal Mining Subsidence Area, Anhui, China. Bull. Environ. Contam. Toxicol. 2019, 103, 10–15. [Google Scholar] [CrossRef]
- Ouyang, Z.; Gao, L.; Yang, C. Distribution, sources and influence factors of polycyclic aromatic hydrocarbon at different depths of the soil and sediments of two typical coal mining subsidence areas in Huainan, China. Ecotoxicol. Environ. Saf. 2018, 163, 255–265. [Google Scholar] [CrossRef]
- Hu, J.; Chen, X.; Chen, Y.; Li, C.; Ren, M.; Jiang, C.; Chen, Y.; An, S.; Xu, Y.; Zheng, L. Nitrate sources and transformations in surface water of a mining area due to intensive mining activities: Emphasis on effects on distinct subsidence waters. J. Environ. Manag. 2021, 298, 113451. [Google Scholar] [CrossRef]
- Ge, Y.; Lou, Y.; Xu, M.; Wu, C.; Meng, J.; Shi, L.; Xia, F.; Xu, Y. Spatial distribution and influencing factors on the variation of bacterial communities in an urban river sediment. Environ. Pollut. 2020, 272, 115984. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Song, Y.; Mao, G.; Lin, B.; Wang, Y.; Gao, G. Spatial variation in bacterial biomass, community composition and driving factors across a eutrophic river. Ecotoxicol. Environ. Saf. 2020, 205, 111113. [Google Scholar] [CrossRef]
- Chen, G.; Wang, X.; Wang, R.; Liu, G. Health risk assessment of potentially harmful elements in subsidence water bodies using a Monte Carlo approach: An example from the Huainan coal mining area, China. Ecotoxicol. Environ. Saf. 2019, 171, 737–745. [Google Scholar] [CrossRef] [PubMed]
- Prosser, J.I. Ecosystem processes and interactions in a morass of diversity. FEMS Microbiol. Ecol. 2012, 81, 507–519. [Google Scholar] [CrossRef]
- Madsen, E.L. Microorganisms and their roles in fundamental biogeochemical cycles. Curr. Opin. Biotechnol. 2011, 22, 456–464. [Google Scholar] [CrossRef]
- Besemer, K.; Luef, B.; Preiner, S.; Eichberger, B.; Agis, M.; Peduzzi, P. Sources and composition of organic matter for bacterial growth in a large European river floodplain system (Danube, Austria). Org. Geochem. 2009, 40, 321–331. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.-P.; Yang, Y.; Niu, Z.-S.; Lu, D.-P.; Zhu, C.-H.; Feng, J.-N.; Wu, J.-Y.; Chen, Y.-R.; Tou, F.-Y.; Liu, M.; et al. Characteristics of microbial community indicate anthropogenic impact on the sediments along the Yangtze Estuary and its coastal area, China. Sci. Total Environ. 2018, 648, 306–314. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Tu, D.; Li, X.; Lu, W.; Li, J. Impact of long-term industrial contamination on the bacterial communities in urban river sediments. BMC Microbiol. 2020, 20, 254. [Google Scholar] [CrossRef]
- Zhang, W.; Lei, M.; Li, Y.; Wang, P.; Wang, C.; Gao, Y.; Wu, H.; Xu, C.; Niu, L.; Wang, L.; et al. Determination of vertical and horizontal assemblage drivers of bacterial community in a heavily polluted urban river. Water Res. 2019, 161, 98–107. [Google Scholar] [CrossRef]
- Zhou, L.; Zhou, Y.; Hu, Y.; Cai, J.; Liu, X.; Bai, C.; Tang, X.; Zhang, Y.; Jang, K.-S.; Spencer, R.G.; et al. Microbial production and consumption of dissolved organic matter in glacial ecosystems on the Tibetan Plateau. Water Res. 2019, 160, 18–28. [Google Scholar] [CrossRef]
- Liu, K.; Liu, Y.; Jiao, N.; Xu, B.; Gu, Z.; Xing, T.; Xiong, J. Bacterial community composition and diversity in Kalakuli, an alpine glacial-fed lake in Muztagh Ata of the westernmost Tibetan Plateau. FEMS Microbiol. Ecol. 2017, 93, 7. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Li, J.; Tang, Y.; Wang, S.; Lu, X.; Cheng, Z.; Zhang, X.; Wu, P.; Chang, X.; Xia, Y. Typhoon-induced turbulence redistributed microplastics in coastal areas and reformed plastisphere community. Water Res. 2021, 204, 117580. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Wang, Y.; Liu, P.; Sun, Y.; Song, Z.; Hu, X. Characteristics of bacterial community structure and function associated with nutrients and heavy metals in coastal aquaculture area. Environ. Pollut. 2021, 275, 116639. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Xu, C.; Zhang, W.; Lin, L.; Wang, L.; Niu, L.; Zhang, H.; Wang, P.; Wang, C. Response of bacterial community in composition and function to the various DOM at river confluences in the urban area. Water Res. 2020, 169, 115293. [Google Scholar] [CrossRef]
- Fan, Y.-Y.; Li, B.-B.; Yang, Z.-C.; Cheng, Y.-Y.; Liu, D.-F.; Yu, H.-Q. Mediation of functional gene and bacterial community profiles in the sediments of eutrophic Chaohu Lake by total nitrogen and season. Environ. Pollut. 2019, 250, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Chen, W.; Zuo, Y.; Lin, L.; Song, L. Heavy metal migration and risk transference associated with cyanobacterial blooms in eutrophic freshwater. Sci. Total Environ. 2018, 613–614, 1324–1330. [Google Scholar] [CrossRef]
- Liu, Y.; Zhu, J.; Gao, W.; Guo, Z.; Xue, C.; Pang, J.; Shu, L. Effects of biochar amendment on bacterial and fungal communities in the reclaimed soil from a mining subsidence area. Environ. Sci. Pollut. Res. 2019, 26, 34368–34376. [Google Scholar] [CrossRef]
- Li, L.; Li, T.; Meng, H.; Xie, Y.; Zhang, J.; Hong, J. Effects of Seven-Year Fertilization Reclamation on Bacterial Community in a Coal Mining Subsidence Area in Shanxi, China. Int. J. Environ. Res. Public Health 2021, 18, 12504. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Li, Y.; Wei, Y.; Meng, H.; Cao, Y.; Lead, J.R.; Hong, J. Effects of fertilization and reclamation time on soil bacterial communities in coal mining subsidence areas. Sci. Total Environ. 2020, 739, 139882. [Google Scholar] [CrossRef]
- Zhu, C.; Zhang, J.; Nawaz, M.Z.; Mahboob, S.; Al-Ghanim, K.A.; Khan, I.A.; Lu, Z.; Chen, T. Seasonal succession and spatial distribution of bacterial community structure in a eutrophic freshwater Lake, Lake Taihu. Sci. Total Environ. 2019, 669, 29–40. [Google Scholar] [CrossRef]
- Zhang, L.; Cheng, Y.; Gao, G.; Jiang, J. Spatial-Temporal Variation of Bacterial Communities in Sediments in Lake Chaohu, a Large, Shallow Eutrophic Lake in China. Int. J. Environ. Res. Public Health 2019, 16, 3966. [Google Scholar] [CrossRef] [PubMed]
- Du, S.; Dini-Andreote, F.; Zhang, N.; Liang, C.; Yao, Z.; Zhang, H.; Zhang, D. Divergent Co-occurrence Patterns and Assembly Processes Structure the Abundant and Rare Bacterial Communities in a Salt Marsh Ecosystem. Appl. Environ. Microbiol. 2020, 86, e00322-20. [Google Scholar] [CrossRef] [PubMed]
- Jiao, C.; Zhao, D.; Zeng, J.; Guo, L.; Yu, Z. Disentangling the seasonal co-occurrence patterns and ecological stochasticity of planktonic and benthic bacterial communities within multiple lakes. Sci. Total Environ. 2020, 740, 140010. [Google Scholar] [CrossRef] [PubMed]
- Hou, F.; Zhang, H.; Xie, W.; Zhou, X.; Zhu, X.; Zhang, D. Co-occurrence patterns and assembly processes of microeukaryotic communities in an early-spring diatom bloom. Sci. Total Environ. 2020, 711, 134624. [Google Scholar] [CrossRef]
- Luo, Z.; Ma, J.; Chen, F.; Li, X.; Zhang, Q.; Yang, Y. Adaptive Development of Soil Bacterial Communities to Ecological Processes Caused by Mining Activities in the Loess Plateau, China. Microorganisms 2020, 8, 477. [Google Scholar] [CrossRef]
- Zhang, L.; Fang, W.; Li, X.; Gao, G.; Jiang, J. Linking bacterial community shifts with changes in the dissolved organic matter pool in a eutrophic lake. Sci. Total Environ. 2020, 719, 137387. [Google Scholar] [CrossRef]
- Li, Y.; Wu, H.; Shen, Y.; Wang, C.; Wang, P.; Zhang, W.; Gao, Y.; Niu, L. Statistical determination of crucial taxa indicative of pollution gradients in sediments of Lake Taihu, China. Environ. Pollut. 2019, 246, 753–762. [Google Scholar] [CrossRef]
- Hu, A.; Ju, F.; Hou, L.; Li, J.; Yang, X.; Wang, H.; Mulla, S.I.; Sun, Q.; Bürgmann, H.; Yu, C.-P. Strong impact of anthropogenic contamination on the co-occurrence patterns of a riverine microbial community. Environ. Microbiol. 2017, 19, 4993–5009. [Google Scholar] [CrossRef]
- Yan, Z.; Hao, Z.; Wu, H.; Jiang, H.; Yang, M.; Wang, C. Co-occurrence patterns of the microbial community in polycyclic aromatic hydrocarbon-contaminated riverine sediments. J. Hazard. Mater. 2019, 367, 99–108. [Google Scholar] [CrossRef]
- Jiao, S.; Liu, Z.; Lin, Y.; Yang, J.; Chen, W.; Wei, G. Bacterial communities in oil contaminated soils: Biogeography and co-occurrence patterns. Soil Biol. Biochem. 2016, 98, 64–73. [Google Scholar] [CrossRef]
- Barberán, A.; Bates, S.T.; Casamayor, E.O.; Fierer, N. Using network analysis to explore co-occurrence patterns in soil microbial communities. ISME J. 2012, 6, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Milici, M.; Deng, Z.-L.; Tomasch, J.; Decelle, J.; Wos-Oxley, M.L.; Wang, H.; Jáuregui, R.; Plumeier, I.; Giebel, H.-A.; Badewien, T.H. Co-Occurrence Analysis of Microbial Taxa in the Atlantic Ocean Reveals High Connectivity in the Free-Living Bacterioplankton. Front. Microbiol. 2016, 7, 649. [Google Scholar] [CrossRef] [PubMed]
- Cram, J.A.; Xia, L.C.; Needham, D.M.; Sachdeva, R.; Sun, F.; Fuhrman, J.A. Cross-depth analysis of marine bacterial networks suggests downward propagation of temporal changes. ISME J. 2015, 9, 2573–2586. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zheng, L.; Wei, X.; Dong, X.; Chen, X. Eutrophication Characteristics and Sources of Nitrogen in Surface Water in Huaibei Mining Area, Anhui. Adm. Tech. Environ. Monit. 2021, 02, 1006–2009. [Google Scholar]
- Chen, X.; Zheng, L.; Jiang, C.; Huang, W.; Dong, X. Characteristics of Surface Water Chemistry and Sulfur Hydrogen Oxygen Isotope Composition in Linhuan Mining Area of Huaibei City, Anhui Province. Earth Environ. 2019, 47, 9. [Google Scholar]
- Kong, L.; Jiang, C.; Zheng, L.; Cheng, H.; Ren, M.; Min, F.; Fang, L. Characters of Hydrochemistry and Their Influenced Factors of Different Waters in the Linhuan Coal Mining Subsidence Area of Huaibei City. J. Lake Sci. 2017, 29, 10. [Google Scholar]
- Jin, X.C.; Tu, Q.Y. The Standard Methods for Observation and Analysis in Lake Eutrophication; Chinese Environment Science Press: Beijing, China, 1990; p. 240. [Google Scholar]
- Fadrosh, D.W.; Ma, B.; Gajer, P.; Sengamalay, N.; Ott, S.; Brotman, R.M.; Ravel, J. An improved dual-indexing approach for multiplexed 16S rRNA gene sequencing on the Illumina MiSeq platform. Microbiome 2014, 2, 6. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, Interactive, Scalable and Extensible Microbiome Data Science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Callahan, B.J.; Mcmurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [PubMed]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA Ribosomal RNA Gene Database Project: Improved Data Processing and Web-Based Tools. Nucleic Acids Res. 2012, 41, D590–D596. [Google Scholar] [CrossRef]
- Liu, J.; He, X.-X.; Lin, X.-R.; Chen, W.-C.; Zhou, Q.-X.; Shu, W.-S.; Huang, L.-N. Ecological Effects of Combined Pollution Associated with E-Waste Recycling on the Composition and Diversity of Soil Microbial Communities. Environ. Sci. Technol. 2015, 49, 6438–6447. [Google Scholar] [CrossRef] [PubMed]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.I. PICRUSt2: An Improved and Extensible Approach for Metagenome Inference. BioRxiv 2019, 672295. [Google Scholar] [CrossRef]
- Yang, Y.; Chen, J.; Chen, X.; Jiang, Q.; Liu, Y.; Xie, S. Cyanobacterial bloom induces structural and functional succession of microbial communities in eutrophic lake sediments. Environ. Pollut. 2021, 284, 117157. [Google Scholar] [CrossRef] [PubMed]
- Csardi, G.; Nepusz, T. The Igraph Software Package for Complex Network Research. InterJournal Complex Syst. 2006, 1695, 1–9. [Google Scholar]
- Zhang, L.; Fang, W.; Li, X.; Lu, W.; Li, J. Strong linkages between dissolved organic matter and the aquatic bacterial community in an urban river. Water Res. 2020, 184, 116089. [Google Scholar] [CrossRef]
- Tang, X.; Xie, G.; Shao, K.; Tian, W.; Gao, G.; Qin, B. Aquatic Bacterial Diversity, Community Composition and Assembly in the Semi-Arid Inner Mongolia Plateau: Combined Effects of Salinity and Nutrient Levels. Microorganisms 2021, 9, 208. [Google Scholar] [CrossRef] [PubMed]
- Zwirglmaier, K.; Keiz, K.; Engel, M.; Geist, J.; Raeder, U. Seasonal and spatial patterns of microbial diversity along a trophic gradient in the interconnected lakes of the Osterseen Lake District, Bavaria. Front. Microbiol. 2015, 6, 1168. [Google Scholar] [CrossRef]
- Hewson, I.; Vargo, G.A.; Fuhrman, J.A. Bacterial Diversity in Shallow Oligotrophic Marine Benthos and Overlying Waters: Effects of Virus Infection, Containment, and Nutrient Enrichment. Microb. Ecol. 2003, 46, 322–336. [Google Scholar] [CrossRef]
- Hu, Y.; Xie, G.; Jiang, X.; Shao, K.; Tang, X.; Gao, G. The Relationships Between the Free-Living and Particle-Attached Bacterial Communities in Response to Elevated Eutrophication. Front. Microbiol. 2020, 11, 423. [Google Scholar] [CrossRef]
- Fox, J.W. The intermediate disturbance hypothesis should be abandoned. Trends Ecol. Evol. 2013, 28, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Faust, K.; Raes, J. Microbial interactions: From networks to models. Nat. Rev. Microbiol. 2012, 10, 538–550. [Google Scholar] [CrossRef] [PubMed]
- Shiri, F.; Anat, K.; Isacc, M.; Uri, G.; Roded, S.; Eytan, R. The Large-Scale Organization of the Bacterial Network of Ecological Co-Occurrence Interactions. Nucleic Acids Res. 2010, 38, 3857–3868. [Google Scholar]
- Roberto, A.A.; Van Gray, J.B.; Leff, L.G. Sediment bacteria in an urban stream: Spatiotemporal patterns in community composition. Water Res. 2018, 134, 353–369. [Google Scholar] [CrossRef] [PubMed]
- Londono, N.; Donovan, A.R.; Shi, H.; Geisler, M.; Liang, Y. Impact of TiO2 and ZnO nanoparticles on an aquatic microbial community: Effect at environmentally relevant concentrations. Nanotoxicology 2017, 11, 1140–1156. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Liu, Z.; Zhang, C.; Wei, Q.; Zhang, S.; Li, M. Spatial and seasonal variations of sediment bacterial communities in a river-bay system in South China. Appl. Microbiol. Biotechnol. 2021, 105, 1979–1989. [Google Scholar] [CrossRef]
- Chalifour, A.; Walser, J.-C.; Pomati, F.; Fenner, K. Temperature, Phytoplankton Density and Bacteria Diversity Drive the Biotransformation of Micropollutants in a Lake Ecosystem. Water Res. 2021, 202, 117412. [Google Scholar] [CrossRef]
- Shapovalova, A.A.; Khijniak, T.V.; Tourova, T.P.; Muyzer, G.; Sorokin, D.Y. Heterotrophic denitrification at extremely high salt and pH by haloalkaliphilic Gammaproteobacteria from hypersaline soda lakes. Extremophiles 2008, 12, 619–625. [Google Scholar] [CrossRef] [PubMed]
- Allers, E.; Gómez-Consarnau, L.; Pinhassi, J.; Gasol, J.M.; Šimek, K.; Pernthaler, J. Response of Alteromonadaceae and Rhodobacteriaceae to Glucose and Phosphorus Manipulation in Marine Mesocosms. Environ. Microbiol. 2007, 9, 2417–2429. [Google Scholar] [CrossRef] [PubMed]
- Kuang, T.; He, A.; Lin, Y.; Huang, X.; Liu, L.; Zhou, L. Comparative analysis of microbial communities associated with the gill, gut, and habitat of two filter-feeding fish. Aquac. Rep. 2020, 18, 100501. [Google Scholar] [CrossRef]
- Zhu, C.-Z.; Li, D.; Chen, W.-J.; Ban, S.-N.; Liu, T.; Wen, H.; Jiang, M. Effects of dietary host-associated Lactococcus lactis on growth performance, disease resistance, intestinal morphology and intestinal microbiota of mandarin fish. Aquaculture 2021, 540, 736702. [Google Scholar] [CrossRef]
- Wang, J.; Feng, J.; Liu, S.; Cai, Z.; Song, D.; Yang, L.; Nie, G. The probiotic properties of different preparations using Lactococcus lactis Z-2 on intestinal tract, blood and hepatopancreas in Cyprinus carpio. Aquaculture 2021, 543, 736911. [Google Scholar] [CrossRef]
- Hou, D.; Zhang, P.; Zhang, J.; Zhou, Y.; Yang, Y.; Mao, Q.; Tsang, D.C.W.; Núñez-Delgado, A.; Luo, L. Spatial variation of sediment bacterial community in an acid mine drainage contaminated area and surrounding river basin. J. Environ. Manag. 2019, 251, 109542. [Google Scholar] [CrossRef]
- Wang, X.; Wang, Y.; Gao, L.; Zhou, X.; Zhao, H.; He, J.; Chen, J.; Wu, Q. Analysis of Physical and Chemical Characteristics of Coal Gangue and Ecological Risk Assessment in Grassland Coal Mine Area. Coal Sci. Technol. 2021, 1–9. [Google Scholar]
- Zhou, L.; Zhou, Y.; Tang, X.; Zhang, Y.; Jang, K.-S.; Székely, A.J.; Jeppesen, E. Resource aromaticity affects bacterial community successions in response to different sources of dissolved organic matter. Water Res. 2020, 190, 116776. [Google Scholar] [CrossRef] [PubMed]
- Bauer, M.; Kube, M.; Teeling, H.; Richter, M.; Lombardot, T.; Allers, E.; Würdemann, C.A.; Quast, C.; Kuhl, H.; Knaust, F.; et al. Whole genome analysis of the marine Bacteroidetes ‘Gramella forsetii’ reveals adaptations to degradation of polymeric organic matter. Environ. Microbiol. 2006, 8, 2201–2213. [Google Scholar] [CrossRef] [PubMed]
- Müllner, A.N.; Schagerl, M. Abundance and Vertical Distribution of the Phytobenthic Community within a Pool and Riffle Sequence of an Alpine Gravel Stream. Int. Rev. Hydrobiol. 2003, 88, 243–254. [Google Scholar] [CrossRef]
- Xie, L.; Xie, P. Long-term (1956–1999) dynamics of phosphorus in a shallow, subtropical Chinese lake with the possible effects of cyanobacterial blooms. Water Res. 2002, 36, 343–349. [Google Scholar] [CrossRef]
- Luo, X.; Xiang, X.; Yang, Y.; Huang, G.; Fu, K.; Che, R.; Chen, L. Seasonal effects of river flow on microbial community coalescence and diversity in a riverine network. FEMS Microbiol. Ecol. 2020, 96, fiaa132. [Google Scholar] [CrossRef]
- Osman, O.A.; Beier, S.; Grabherr, M.; Bertilsson, S. Interactions of Freshwater Cyanobacteria with Bacterial Antagonists. Appl. Environ. Microbiol. 2017, 83, e02634-16. [Google Scholar] [CrossRef] [PubMed]
- Cai, W.; Li, Y.; Shen, Y.; Wang, C.; Wang, P.; Wang, L.; Niu, L.; Zhang, W. Vertical distribution and assemblages of microbial communities and their potential effects on sulfur metabolism in a black-odor urban river. J. Environ. Manag. 2019, 235, 368–376. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Chen, H.; Yang, J.R.; Liu, M.; Huang, B.; Yang, J. Distinct Patterns and Processes of Abundant and Rare Eukaryotic Plankton Communities Following a Reservoir Cyanobacterial Bloom. ISME J. Emultidisciplinary J. Microb. Ecol. 2018, 12, 2263–2277. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Pan, J.; Yang, J.; Zhou, Z.; Pan, Y.; Li, M. Patterns and processes of free-living and particle-associated bacterioplankton and archaeaplankton communities in a subtropical river-bay system in South China. Limnol. Oceanogr. 2020, 65, S161–S179. [Google Scholar] [CrossRef]
- Xun, W.; Liu, Y.; Li, W.; Ren, Y.; Xiong, W.; Xu, Z.; Zhang, N.; Miao, Y.; Shen, Q.; Zhang, R. Specialized metabolic functions of keystone taxa sustain soil microbiome stability. Microbiome 2021, 9, 35. [Google Scholar] [CrossRef] [PubMed]
- Krause, A.E.; Frank, K.A.; Mason, D.M.; Ulanowicz, R.E.; Taylor, W.W. Compartments revealed in food-web structure. Nature 2003, 426, 282–285. [Google Scholar] [CrossRef]
- Coyte, K.Z.; Schluter, J.; Foster, K.R. The ecology of the microbiome: Networks, competition, and stability. Science 2015, 350, 663–666. [Google Scholar] [CrossRef]
- Xun, W.; Yan, R.; Ren, Y.; Jin, D.; Xiong, W.; Zhang, G.; Cui, Z.; Xin, X.; Zhang, R. Grazing-induced microbiome alterations drive soil organic carbon turnover and productivity in meadow steppe. Microbiome 2018, 6, 170. [Google Scholar] [CrossRef]
- Dong, Y.; Gao, J.; Wu, Q.; Ai, Y.; Huang, Y.; Wei, W.; Sun, S.; Weng, Q. Co-occurrence pattern and function prediction of bacterial community in Karst cave. BMC Microbiol. 2020, 20, 137. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Walder, F.; Büchi, L.; Meyer, M.; Held, A.Y.; Gattinger, A.; Keller, T.; Charles, R.; van der Heijden, M.G.A. Agricultural intensification reduces microbial network complexity and the abundance of keystone taxa in roots. ISME J. 2019, 13, 1722–1736. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Schlaeppi, K.; Van Der Heijden, M.G.A. Keystone taxa as drivers of microbiome structure and functioning. Nat. Rev. Microbiol. 2018, 16, 567–576. [Google Scholar] [CrossRef]
- Zhang, L.; Guo, B.; Zhang, Q.; Florentino, A.; Xu, R.; Zhang, Y.; Liu, Y. Co-digestion of blackwater with kitchen organic waste: Effects of mixing ratios and insights into microbial community. J. Clean. Prod. 2019, 236, 117703. [Google Scholar] [CrossRef]
- Fredrickson, J.K.; Romine, M.F.; Beliaev, A.S.; Auchtung, J.M.; Driscoll, M.E.; Gardner, T.S.; Nealson, K.H.; Osterman, A.L.; Pinchuk, G.; Reed, J.L.; et al. Towards environmental systems biology of Shewanella. Nat. Rev. Microbiol. 2008, 6, 592–603. [Google Scholar] [CrossRef] [PubMed]
- Xie, G.; Tang, X.; Shao, K.; Zhu, G.; Gao, G. Bacterial diversity, community composition and metabolic function in Lake Tianmuhu and its dammed river: Effects of domestic wastewater and damming. Ecotoxicol. Environ. Saf. 2021, 213, 112069. [Google Scholar] [CrossRef] [PubMed]
- Yan, Q.; Bi, Y.; Deng, Y.; He, Z.; Wu, L.; van Nostrand, J.D.; Shi, Z.; Li, J.; Wang, X.; Hu, Z.; et al. Impacts of the Three Gorges Dam on microbial structure and potential function. Sci. Rep. 2015, 5, 8605. [Google Scholar] [CrossRef]
- Luo, J.; Tan, X.; Liu, K.; Lin, W. Survey of sulfur-oxidizing bacterial community in the Pearl River water using soxB, sqr, and dsrA as molecular biomarkers. 3 Biotech. 2018, 8, 73. [Google Scholar] [CrossRef] [PubMed]
- Parker, J.L.; Shaw, J.G. Aeromonas spp. clinical microbiology and disease. J. Infect. 2011, 62, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Vendrell, D.; Balcázar, J.L.; Ruiz-Zarzuela, I.; de Blas, I.; Gironés, O.; Múzquiz, J.L. Lactococcus garvieae in fish: A review. Comp. Immunol. Microbiol. Infect. Dis. 2006, 29, 177–198. [Google Scholar] [CrossRef]
- Wu, J.; Liu, M.; Zhou, M.; Wu, L.; Yang, H.; Huang, L.; Chen, C. Isolation and genomic characterization of five novel strains of Erysipelotrichaceae from commercial pigs. BMC Microbiol. 2021, 21, 125. [Google Scholar] [CrossRef] [PubMed]
- Nematollahi, A.; Decostere, A.; Pasmans, F.; Haesebrouck, F. Flavobacterium psychrophilum infections in salmonid fish. J. Fish Dis. 2003, 26, 563–574. [Google Scholar] [CrossRef] [PubMed]
- Paczosa, M.K.; Mecsas, J. Klebsiella pneumoniae: Going on the Offense with a Strong Defense. Microbiol. Mol. Biol. Rev. 2016, 80, 629–661. [Google Scholar] [CrossRef]
- Xu, Z.; Te, S.H.; He, Y.; Gin, K.Y.-H. The Characteristics and Dynamics of Cyanobacteria–Heterotrophic Bacteria Between Two Estuarine Reservoirs—Tropical Versus Sub-Tropical Regions. Front. Microbiol. 2018, 9, 2531. [Google Scholar] [CrossRef]
- Ruprecht, J.E.; Birrer, S.C.; Dafforn, K.A.; Mitrovic, S.M.; Crane, S.L.; Johnston, E.L.; Wemheuer, F.; Navarro, A.; Harrison, A.J.; Turner, I.L.; et al. Wastewater effluents cause microbial community shifts and change trophic status. Water Res. 2021, 200, 117206. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Luo, G.; Guo, J.; Xiao, Y.; Zhang, F.; Guo, S.; Ling, N.; Shen, Q. Microbial generalist or specialist: Intraspecific variation and dormancy potential matter. Mol. Ecol. 2021, 31, 161–173. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fan, T.; Fang, W.; Zhao, Y.; Lu, A.; Wang, S.; Wang, X.; Xu, L.; Wei, X.; Zhang, L. Spatial Variations of Aquatic Bacterial Community Structure and Co-Occurrence Patterns in a Coal Mining Subsidence Lake. Diversity 2022, 14, 674. https://doi.org/10.3390/d14080674
Fan T, Fang W, Zhao Y, Lu A, Wang S, Wang X, Xu L, Wei X, Zhang L. Spatial Variations of Aquatic Bacterial Community Structure and Co-Occurrence Patterns in a Coal Mining Subsidence Lake. Diversity. 2022; 14(8):674. https://doi.org/10.3390/d14080674
Chicago/Turabian StyleFan, Tingyu, Wangkai Fang, Yifan Zhao, Akang Lu, Shun Wang, Xingming Wang, Liangji Xu, Xiangping Wei, and Lei Zhang. 2022. "Spatial Variations of Aquatic Bacterial Community Structure and Co-Occurrence Patterns in a Coal Mining Subsidence Lake" Diversity 14, no. 8: 674. https://doi.org/10.3390/d14080674
APA StyleFan, T., Fang, W., Zhao, Y., Lu, A., Wang, S., Wang, X., Xu, L., Wei, X., & Zhang, L. (2022). Spatial Variations of Aquatic Bacterial Community Structure and Co-Occurrence Patterns in a Coal Mining Subsidence Lake. Diversity, 14(8), 674. https://doi.org/10.3390/d14080674