1. Introduction
Mithun (
Bos frontalis), also known as ‘Gayal’, and ‘Cattle of the mountain’, an endangered ruminant species belonging to the family Bovidae, is indigenous to the eastern Himalayas. It plays an important role in the traditions and rituals of the vast tribal population of this North-Eastern Hill (NEH) region of India, being the pride of northeast India. There are various hypotheses on the origin of mithun reported in the literature. Mithun was first described as ‘the Ceremonial Ox’ of the Naga tribes of India, and the Indo-Myanmar border was reported to be the place of origin of mithun [
1]. Three possible hypotheses regarding the origin of mithun were proposed, namely, (i) directly domesticated from wild Gaur/Indian Bison (
Bos gaurus) [
2,
3,
4,
5,
6,
7], (ii) hybrid decent from the crossing of wild Gaur and domestic cattle [
8,
9,
10], and (iii) crossing with a wild Bos, which is now extinct [
11,
12]. Mithun is mainly reared as a meat animal and its meat (meef) is relished among native tribes because of its unique texture, taste, and flavor. Similarly, milk obtained from mithun cows is reported to be superior in terms of quality over the milk of other bovines, and contains double the quantity of total energy value as compared to traditional cow milk [
13,
14].
India has the largest mithun population (~97.57%) in the world (0.38 million) [
15], while Myanmar, Bangladesh, China, and Bhutan account for approximately 3000 (0.96%), 1000 (0.32%,), 3000 (0.96%), and 570 (0.18%) respectively [
16]. The dwindling mithun population has made this unique species classified as an endangered species by the International Union for Conservation of Nature and Natural Resources (
www.iucnredlist.org/) [
10].
The wild gaur aka Indian Bison (
Bos gaurus), the largest among the bovine family, is a highly endangered species in the wild and declared vulnerable by the International Union for Conservation of Nature and Natural Resources (IUCN) since 1986 (
www.iucnredlist.org/, accessed on 10 January 2022) due to the rapidly declining population [
17]. It is a massively built animal and is very strong. These are kept in various Indian zoos and protected National Parks as a part of conservation efforts. Recently, Sri Chamarajendra Zoological Gardens, Mysuru (Mysore), India is in the process of initiating an ambitious breeding and conservation program for wild gaurs. This necessitates the investigation of population structure and genetic diversity present in wild gaur population. Tho-tho is one of the indigenous cattle breeds, available mostly in Kohima and Phek district of Nagaland, and used by local tribal people for meat and dung purposes. It is a small- to medium-sized animal (adult weighing 260–300 kg) with pure black or black with white spot coat [
18]. It is used for draught, manure, and meat purposes (
https://nbagr.icar.gov.in/en/thutho-cattle/, accessed on 9 April 2022).
Microsatellites (Simple Sequence Repeats, SSRs) have been widely used for the genetic characterization and diversity analysis for livestock worldwide and have been recommended by the FAO as microsatellites are highly informative, codominant, highly specific, transferable among related species, and have relatively low costs. Therefore, microsatellites continue to be widely used, especially for various wild species [
19,
20,
21,
22]. SSRs have been the most frequently used genetic marker in population genetics in the past two decades [
23]. Recently, Single Nucleotide Polymorphic loci (SNPs) have been used in a variety of genetic studies [
24], but they could not replace SSRs completely, even in the genomic era, due to their potential benefits [
25]. Multi-allelic nature and high polymorphism of SSR markers help to establish the relationship among the individuals even with fewer markers. SSR markers are immensely valuable in studies of variation detection, diversity analysis, phylogeny, population structure, gene mapping, and association studies in vertebrates [
26,
27,
28,
29] and fish species [
30].
Several studies have demonstrated the successful transferability and applicability of gene-based microsatellite markers across related genera in both crop plants [
31] and livestock species, particularly recommending the use of cattle (
Bos taurus) microsatellite markers for population genetic analysis for other related Bos species for which microsatellites have not been developed, namely
Bos frontalis and
Bos gaurus, respectively [
32,
33,
34,
35,
36,
37]. It was decided to test microsatellites markers that have been developed for cattle [
26,
38], to assess the population structure and genetic diversity of mithun and gaur (Indian bison) population, being in the same Bos family. Furthermore, mtDNA based studies provides additional support and is regarded as an informational tool for genetic diversity, evolutionary studies due to the near-neutrality, maternal inheritance, and clock-like nature of its substitution rate [
39]. The Displacement region (D-loop) proves to be a particularly useful genetic marker because it evolves much faster than the coding region of the mtDNA [
40]. Hence, studying mtDNA becomes a quintessential tool to explore population structures and infer evolutionary histories.
Even if all these studies showed high suitability of bovine microsatellite markers for genetic characterization of domesticated mithun and wild gaur, as a member of the subfamily Bovinae, information on the genetic diversity of Indian mithuns and wild gaurs are scanty so far. Thus, it is important to determine the present level of genetic diversity and population structure in the mithun and gaur population to aid the development of suitable breeding strategies and propose well-versed recommendations to support their ongoing conservation and genetic improvement program. This is probably the first attempt, to our knowledge, for the genetic characterization of Indian mithun, Indian Bison/wild gaurs, and Tho-tho cattle using microsatellite (SSR) markers.
Therefore, the present study was performed to assess the levels of genetic diversity and population structure among mithun, wild gaur, and Tho-tho cattle. The results will help in a formulating effective breeding policy and shaping future conservation plans, as these sub-species come under endangered risk category. There is a need for improvement and at the same time maintaining subspecies purity by reducing the possible admixture due to subspecies hybridization.
2. Materials and Methods
2.1. Samples and DNA Extraction
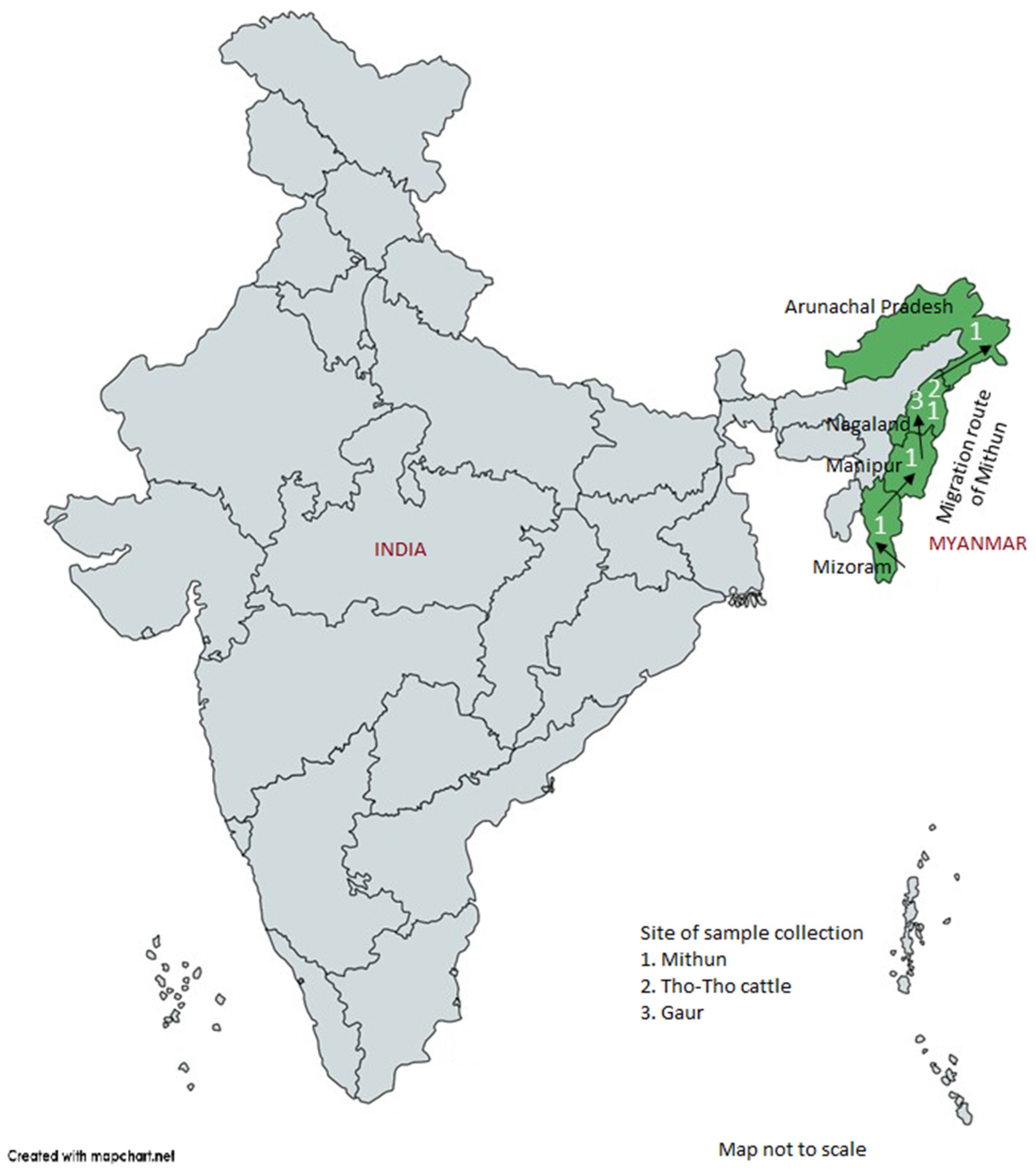
A total of 163 random blood samples from genetically unrelated four mithun populations, corresponding to four North-Eastern Indian states (
Figure 1), namely, Arunachal Pradesh (
n = 40), Nagaland (
n = 41), Manipur (
n = 16), and Mizoram (
n = 66); additionally, 10 blood samples from Indian bison/wild gaur (
Bos gaurus) from Mysore Zoo, Karnataka State, India were collected for diversity analysis. Twenty-five samples of a local cattle breed, Tho-tho, were also collected for positive control and comparative purpose. Genomic DNA from each sample was isolated from white blood cells by using a Promega DNA isolation kit (Promega, Madison, WI, USA) [
41] and stored at −20 °C until further use.
2.2. Microsatellite Loci
Thirty cattle microsatellite loci (SSR markers) from the FAO (1996) MoDAD list [
38], mapped to 20 different autosomes (
Table 1), were tested for cross-species amplification and analysis of genetic diversity in the Indian mithun and wild gaur populations. Primer pairs for microsatellite markers were synthesized by Applied Biosystem (Life technologies, CA, USA). The forward primers of each locus were 5′ end-labeled, with either FAM, VIC, NED, or PET fluorescent tag, whereas the reverse primers were unlabeled. Polymerase chain reactions (PCR) were carried out in a 50 µL reaction volume containing 1.5 mm MgCl
2, 10 mM dNTPs, 100 ng of each primer, 100 ng of template DNA, and 3U of Fast Taq Polymerase (Chromous Biotech, Bangalore, India).
2.3. Mitochondrial DNA Sequencing
The transmembrane protein coding cytochrome B gene (CYTB) was amplified by PCR, using primer pair L14724 (5′-CGAAGCTTGATATGAAAAACCATCGTTG-3′) and H15915R (5′-GGAATTCATCTCTCCGGTTTACAAGAC-3′) as described by [
42].
These two primers yielded a PCR product of 1246 bp, representing the complete CYTB gene of mithun (GenBank: JQ404407.1).
2.4. Multiplex PCR and Genotyping
All the 30 microsatellite markers were initially tested for amplification of genomic DNA from a panel of 20 Indian mithuns and five gaurs. Genomic DNA isolated from eight local Tho-tho cattle (
Bos indicus) were used as positive control. In total, 19 out of 30 cattle microsatellite markers successfully amplified mithun and gaur genomic DNA. Subsequently, all the genomic DNA samples were amplified using these 19 sets of microsatellite markers in four multiplex PCR set-ups (
Table 2).
Amplification was performed in ABI Veriti thermal cycler (Applied Biosystems, Switzerland). The capillary electrophoresis was performed in an ABI Genetic Analyzer 3500xL (Applied Biosystem, CA, USA), according to manufacturer’s recommendations. The subsequent gel analysis and fragment sizing were performed with GENEMAPPER® version 4.1 software (Applied Biosystems, Waltham, MA, USA) with LIZ 500R size standard (Applied Biosystems, Switzerland). All the 19 amplified products for each microsatellite locus were cloned in T-Vector, the plates were screened for clones, and the positive clones were sequenced to analyze for presence of number of two-base repeats.
Polymerase Chain Reaction (PCR) was carried out on about 50 ng genomic DNA in a 25 μL reaction volume using Thermal Cycler (Applied Biosystem, Waltham, MA, USA). The reaction mixture consisted of 200 μM of each dNTPs, 1.5 mM MgCl2, 50 pmol primer, 0.5 U Taq polymerase (Promega, USA), and Taq buffer. Negative controls (lacking template DNA) were included in all reactions, and produced no products. The PCR reaction cycle was accomplished by denaturation for 6 min at 94 °C, 30 cycles of 94 °C for 45 s, 60 °C for 30 s, 72 °C for 60 s, and finally extension at 72 °C for 6 min, before cooling to 4 °C for 10 min. The size of amplification product was checked by loading 10 μL PCR product onto 2% agarose gel containing 0.5 μL/mL ethidium bromide. The product was purified using a QIA quick PCR purification kit (Qiagen, Hilden, Germany). Purified product was labeled using the Big Dye Terminator 3.1 Cycle sequencing kit (Applied Biosystems, Foster City, CA, USA) and sequenced directly using an ABI3100 Prism automatic DNA sequencer following manufacturer instructions. The primers used for sequencing were the same as those used in the PCR. Both strands of PCR product were completely sequenced. All final sequences were determined from both strands for verification.
2.5. Statistical Data Analysis
To identify the genetic structure of the given population and assign individuals to populations, the software STRUCTURE version 2.3.4 was used [
43]. To estimate the optimal number of groups (K), STRUCTURE was run with K varying from 1 to 10, with five runs for each K value. To determine the true value of K, ad hoc statistic ΔK was followed. Parameters were set to 100,000 burn-in periods and 500,000 Markov Chain Monte Carlo (MCMC) replications after burn-in with an admixture and allele frequencies correlated model. The method described by [
44] was used to estimate the most probable K value for the analyzed data, using the web tool Structure Harvester ver. 0.6. application [
45].
Different measurements of within breed genetic variations, namely, observed number of alleles (Na), effective number of alleles (Ne), observed heterozygosity (H
o), and Nei’s unbiased expected heterozygosity (H
e) [
46], were estimated using POPGENE software v1.32 [
47]. Polymorphic information content (PIC) for each locus was calculated by the formula given by [
48]:
where,
Pi and
Pj are frequencies of the
ith and
jth alleles, respectively.
The allele data frequencies were used to assess if there was any bottleneck in the mithun population in recent history using BOTTLENECK software v1.2.02 [
49]. The BOTTLENECK program was used to measure the genetic bottleneck through the test of gene diversity excess relative to that of under mutation-drift equilibrium. Reduction in the population genetic signatures was determined by the Wilcoxon’s heterozygosity excess test, the null hypothesis of the test being that all loci are in mutation-drift equilibrium. Three mutation models of microsatellite evolution were considered: Infinite allele model (IAM), Stepwise Mutation Model (SMM), and Two-Phase Model (TPM).
The allelic data were subjected to estimation of genetic distances among genotypes using simple matching coefficients by bootstrapping 1000 times and they were clustered using a neighbor-joining method using Darwin software version 6.0 [
50]. Furthermore, an analysis of molecular variance (AMOVA) was performed to describe variance components among individuals and the population differentiation among the seven assumed subpopulations using GeneAlEx 6.502 program [
51] with 1000 permutations. Principal coordinate analysis (PCoA) was performed to highlight the resolving power of the ordination and the first two components were used to represent the genotypes in the graphical form. PCoA and dissimilarity matrix was performed by using DARwin software version. 6.0. Genetic differentiation among the assumed subpopulation was analyzed using Nei’s gene diversity statistics using GenAlEx program version 6.502.
2.6. Mitochondrial DNA Statistical Analysis
The raw DNA sequences were analyzed manually using EditSeq (DNASTAR/Lasergene 11.0, Madison, Wisconsin, USA) and the online Nucleotide BLAST program (
https://blast.ncbi.nlm.nih.gov/Blast.cgi, accessed on 10 January 2022) was used for alignment to construct consensus sequences. No gaps in the aligned sequences were permitted in the final analysis. We compared the 21 CYTB haplotypes of a 1021 bp fragment of mitochondrial DNA region obtained from 71 bovine samples from India. Diversity parameters including haplotype diversity (Hd), nucleotide diversity (π) within bovine populations, and average number of sequence differences (k) were estimated by Arlequin 3.1 [
52].
3. Results
In total, 19 (63%) out of 30 cattle microsatellite markers (
Table S1) revealed successful amplification patterns in 163 maithuna and 10 gaur samples, while the remaining 11 cattle-specific microsatellite markers failed to yield any amplification. Based on prior information on PCR products’ sizes, 19 microsatellite loci were grouped into four sets, containing five (ETH225, HAUT27, BM1818, LSTS030, and ETH185), four (HEL1, ETH3, BM1824, and ILSTS034), five (MM12, ETH10, HEL13, ILSTS033, and ETH152) and five loci (CSSM66, BM2113, ILSTS006, ILSTS011, and ILSTS054), respectively (
Table 1). Fifteen of the positive clones which were sequenced and analyzed for presence of mithun-specific microsatellite repeats were submitted in GenBank (Accession KF564956-KF564970,
Table 1). Genotype data collected from the 19 amplified microsatellites were used for genetic diversity studies of Indian mithun and wild gaur populations.
3.1. SSR Polymorphism among the Mithun, Gaur, and Tho-tho Population
The number of alleles, allele size, and polymorphism information content (PIC) detected among 163 mithun, 10 gaur, and 25 Tho-tho samples using 19 SSR markers is presented in
Table 1. In the present diversity analysis, the PIC value ranged from 0.30 for HAUT27 to 0.89 for ILSTS006 microsatellite, with an average of 0.63 for mithun. PIC values in gaur population ranged from 0.32 for CSSM66 to 0.82 for LSTS030. In Tho-tho cattle the PIC value ranged 0.43 (ETH3) to 0.88 (CSSM66) (
Table 1). The ILSTS006 and LSTS030 microsatellite showed higher discriminatory power to distinguish genotypes due to its high PIC value. A total of 15 microsatellite (79%) markers out of the 19 microsatellite loci were highly polymorphic and showed high PIC value (>0.50) in both mithun and gaur genotypes. Sixteen markers were highly polymorphic for Tho-tho population.
3.2. Population Genetic Diversity
The individual genetic diversity of mithun and gaur population is presented in
Table 1. A total of 254 and 89 alleles were detected in Mithun and Gaur with allele numbers ranging from 8 to 25 in Mithun and 2 to 7 in gaur. The Tho-tho population comprised 136 alleles with allele number ranging 3 to 12 (
Table 1). Hence, each of the panel of 15 microsatellites—Panel 1 (ETH225, BM1818, ILSTS030, ETH185, HEL1, ETH3, ILSTS034, MM12, HEL13, ILSTS033, ETH152, CSSM66, BM2113, ILSTS006, and ILSTS011) and Panel 2 (HAUT27, BM1818, ILSTS030, ETH185, HEL1, ETH3, BM1824, ILSTS034, MM12, HEL13, ETH152, BM2113, ILSTS006, ILSTS011 and ILSTS054)—from the FAO standard panel for cattle diversity studies appear to be most suitable for diversity studies in Indian mithun and Gaur, respectively, due to their high informativeness. The observed heterozygosity (H
o) ranged from 0.15 to 0.94 in mithun with an average of 0.48 and 0.01 to 0.99 in gaur with an average of 0.62 across all 19 loci, while expected heterozygosity and gene diversity (H
e) ranged from 0.31 to 0.89 in mithun, with an average of 0.66 and 0.36 to 0.83 in gaur with an average of 0.71, respectively, across all 19 loci. The observed and expected heterozygosity for Tho-tho population was 0.33–0.92 and 0.59–0.93, respectively. The average observed and expected heterozygosity was found to be 0.75 and 0.77, respectively, for the Tho-tho population.
The overall observed heterozygosity (H
o) ranged from 0.226 to 0.867 with an average of 0.54 across all 19 loci in the present study, comprising animals from the mithun, gaur, and Tho-tho cattle population. The total observed heterozygosity (0.54) was far lower than the total expected heterozygosity (0.858), which is supported by a low gene flow (Nm) value (
Table 2). Average F
IS and F
IT values were 0.29 and 0.377 (
Table 2), respectively, which is obvious and has indicated less inbreeding in the mithun, gaur, and Tho-tho cattle population due to the cross-bred nature of reproduction. Gene diversity ranged from 0.524 (BM1824) to 0.876 (CSSM66), with an average of 0.745.
3.3. Genetic Relationship among the Germplasm
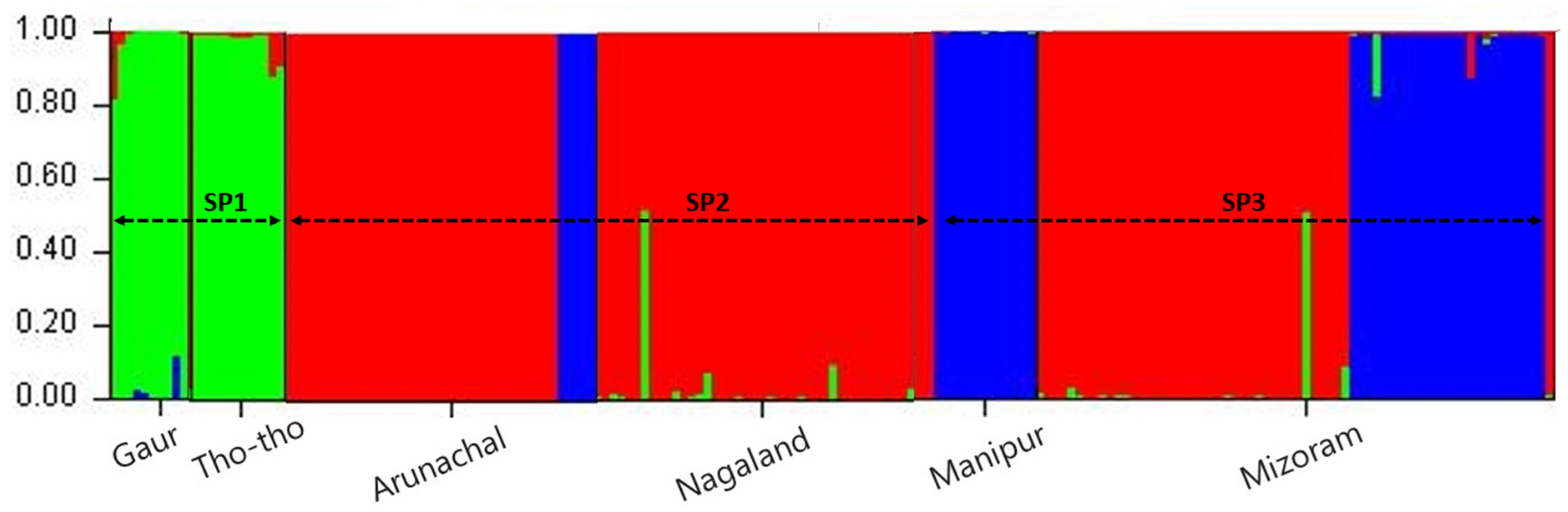
In the present study grouping of mithun, gaur, and Tho-tho population was determined using STRUCTURE analysis. Population structure of the 185 genotypes (163 mithun, 10 Gaur, and 25 Tho-Tho) was analyzed using a Bayesian model-based clustering approach with the k value ranging from 1 to 10 with 10 iterations using 19 polymorphic microsatellite markers data. The ΔK was found higher for K = 3 than other values of K and the standard deviation was least at K = 3. Hence, the true number of the populations was considered as three (SP1, SP2, and SP3), which revealed that in the present study, all the bovine populations (mithun, gaur, and Tho-tho) can be clustered into three subgroups. Genotypes under different subpopulations were categorized as pure or admixture based on the membership fractions. The accessions stratified in a particular subpopulation with the probability of ≥80% were considered as pure and assigned to corresponding subgroups, while <80% were categorized as admixture (
Figure 2).
Based on STRUCTURE analysis, the subpopulation SP1 comprised Tho-tho cattle and gaur. Subpopulation SP2 consists of mithun population from Arunachal Pradesh and Nagaland, while subpopulation SP3 consists of mithun population from Manipur and Mizoram. The distinct subpopulations were obtained as per geographical locations and species.
Among the subpopulations, the SP3 was highly differentiated from SP2 (FST = 0.146) and SP1 (FST = 0.095), followed by differentiation between SP2 and SP1 (FST = 0.080). Subpopulation SP3 consists of mithuns from Manipur and Mizoram. This genetic difference might be attributed to natural selection favoring a different group of alleles in different niches or ecosystems and geographical barriers such as mountains, rivers, and valleys, which leads to low cross breeding or migration of genes/alleles between different subpopulations. This is further supported by moderate gene flow values among subpopulations.
The genetic diversity at subpopulation was studied in terms of the mean number of alleles (Na), number of effective alleles (Ne), Shannon’s information index (I), observed heterozygosity (H
o), gene diversity, unbiased expected heterozygosity (uH
e), and wright’s fixation index (f), which is presented in
Table 3. The Ne, I, and F are comparable among subpopulations. The highest number of effective alleles was present in the SP1 population, which consists of the gaur and Tho-tho cattle population. The lowest number of effective alleles was found in the SP2 population. This indicated that the SP1 population is more stable in the long term and has more buffering genes for wider adaptability. Shannon’s information index varied from 2.20 (SP1) to 1.58 (SP2). An analysis of Shannon’s information index revealed that the SP1 population is different from the SP2 and SP3 populations by showing the highest Shannon’s information index. The highest gene diversity was observed in subpopulation SP1.
3.4. Analysis for Molecular Variance
The three subpopulations obtained from the structure analysis were subjected to AMOVA, to measure the percentage of molecular variance between subpopulations, among individuals and within individuals. AMOVA revealed the presence of 15% of the variation among populations and 29% of variation among individuals, whereas 56% of variation was present within individuals (
Table 4). AMOVA estimates indicated that most of the variation in mithun germplasm is present within individuals. Lower F
IS (0.336) and F
IT (0.436) estimated at subpopulation level (
Table 4) in the entire population indicated the presence of a high amount of heterozygosity because of the cross-bred nature of mithun, gaur, and Tho-tho cattle. The F
ST value of 0.151 (
Table 4) indicates distinctness and presence of genetic variation among the subpopulations of mithuns.
Pairwise F
ST values of the subpopulation range from 0.080 to 0.146 (
Table 5) and showed significant differentiation among all the pairs, which suggested that all the three subpopulation were significantly different from each other. Based on the pairwise F
ST estimate, SP2 and SP3 showed the highest level of differentiation from each other and subpopulation SP1 exhibited less differentiation from SP2 and SP3.
Nei’s genetic distance varied from 0.626 to 1.397. The maximum distance was observed between subpopulation SP1 and SP3, and minimum distance was observed between SP1 and SP2 (
Table 6). This is supported by a low gene flow between SP1 and SP3.
3.5. Neighbor-Joining Based Clustering
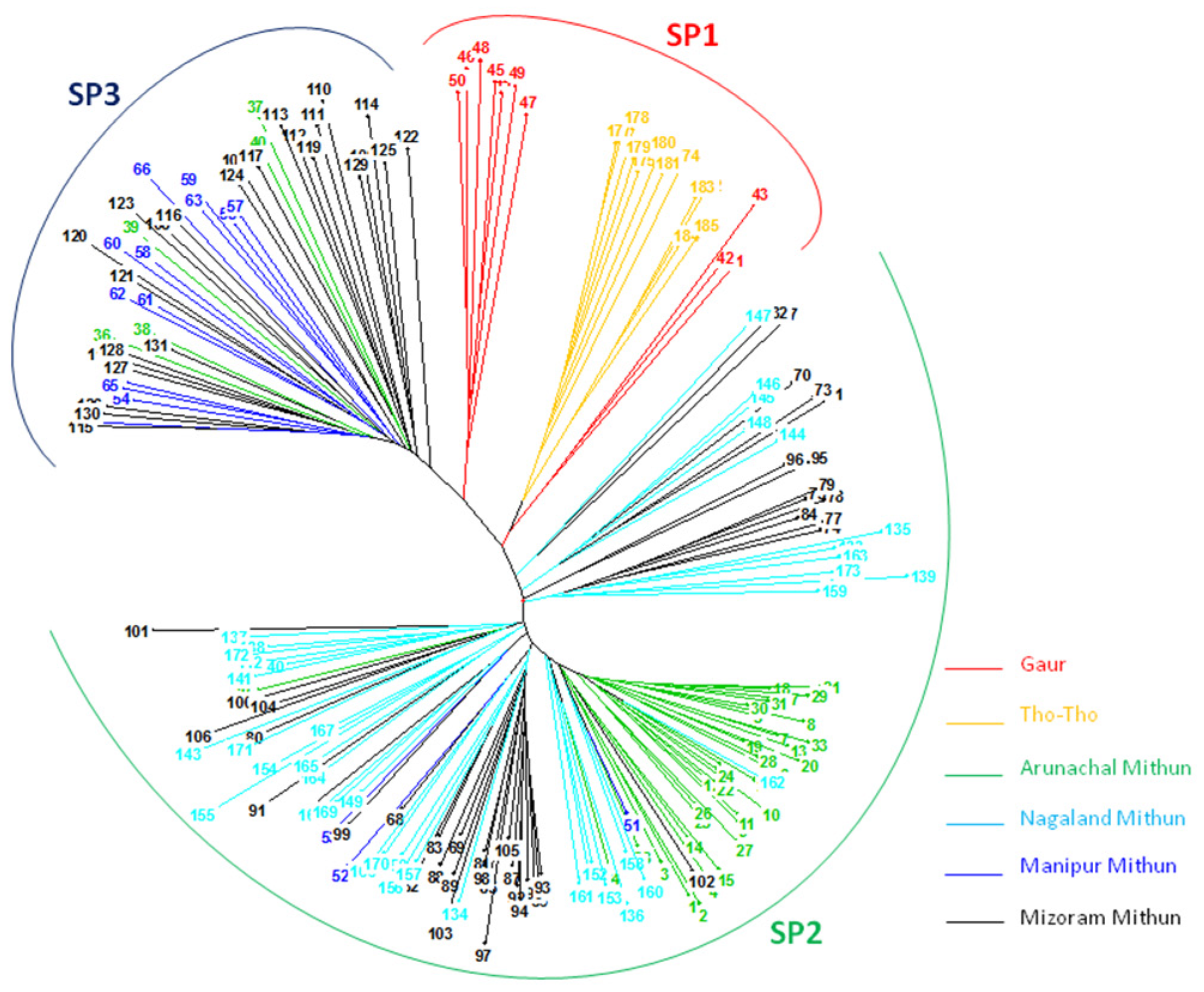
An unrooted neighbor-joining cluster analysis based on unweighted method using the 19 SSR marker allelic data classified the 185 bovines (mithun, guar, and Tho-tho) into three clusters (
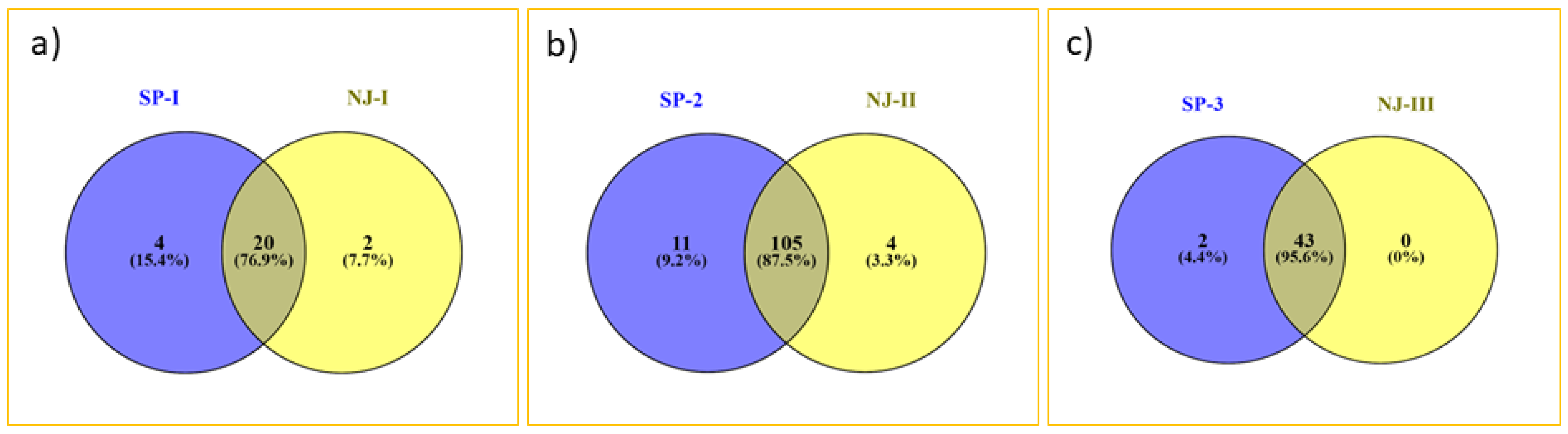
Figure 3). The result of distance-based neighbor-joining cluster analysis is similar to the model-based grouping pattern. Gaur and Tho-tho cattle are grouped adjacent to each other but distinct from each other. Nagaland and Arunachal mithuns were grouped in cluster 2, whereas Manipur and Mizoram mithuns were clustered in cluster 3. Few of Mizoram mithuns have shown close similarity with Nagaland mithuns. This cluster analysis revealed the presence of significant amount of genetic diversity among the bovine cattle population. The results of grouping pattern generated by the model-based STRUCTURE analysis were compared with distance based neighbor-joining cluster analysis using a Venn diagram (
Figure 4 Venn). Cluster-I, based on the neighbor-joining analysis, showed 76.9% of similarity of genotypes with subpopulation SP1 generated through the model-based analysis. Cluster-II exhibited 95.6% of similarity of genotypes with the subpopulation SP2. Similarly, 87.5% of correspondence was observed between Cluster-III and the SP3 subpopulation.
3.6. Principal Component Analysis (PCA)
The PCA based on 19 micro-satellite allelic datapoints determined the genetic relatedness among the bovine cattle population. The first two coordinates of PCA explained the 28.69% of variation and clearly distinguished the three subpopulations obtained from the STRUCTURE analysis. The first coordinate of PCA explained 24.96% of the variation and the second coordinate explained 3.73% of the variation (
Figure S1). The PCA result showed good correspondence with the neighbor-joining clustering and STRUCTURE grouping patterns.
3.7. Principal Coordinate Analysis (PCoA)
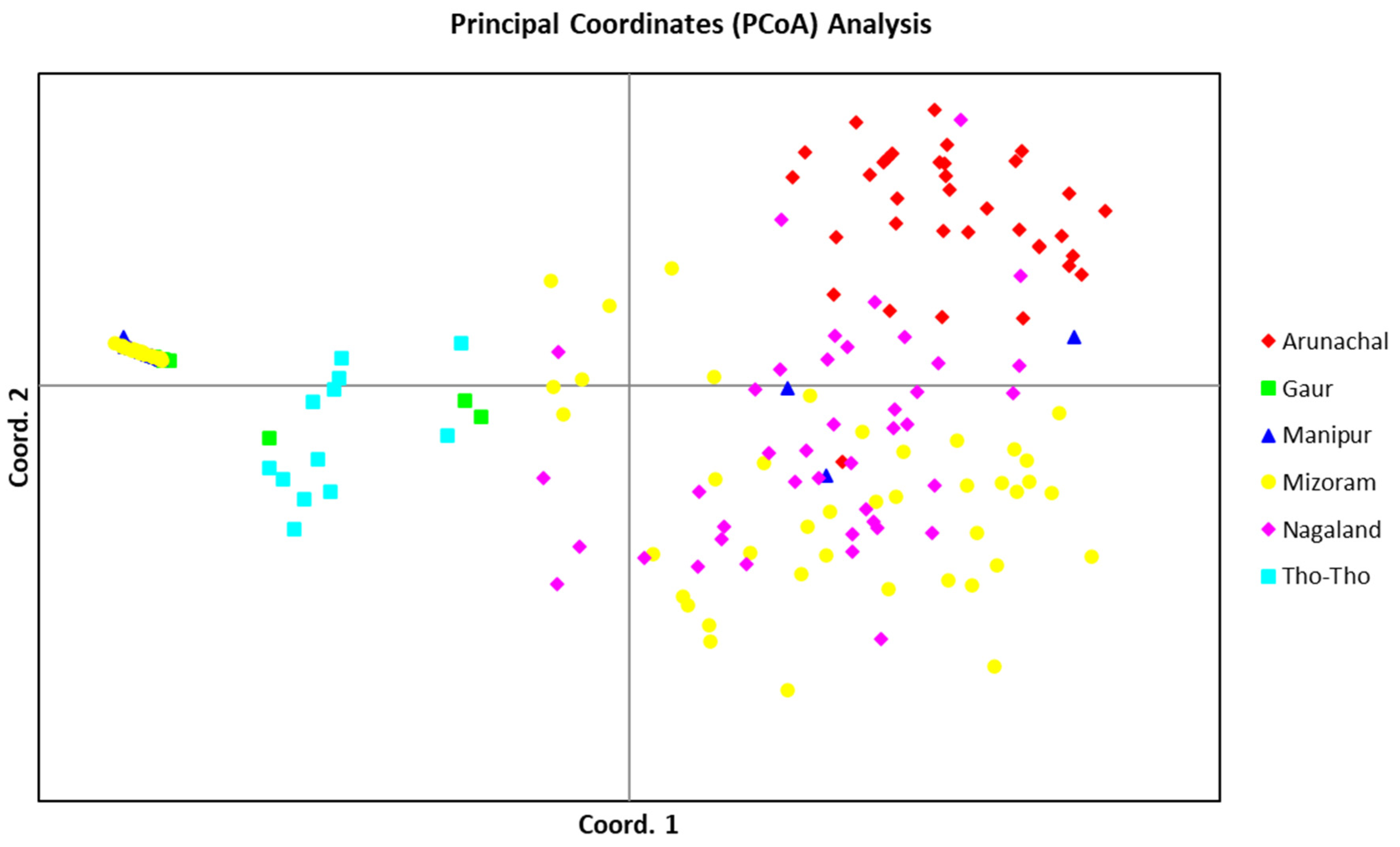
The PCoA, using allelic data of 19 SSR markers, determined the patterns of variation among the three bovine populations (Mithun, Gaur, and Tho-Tho). The PCoA analysis also clearly separated the STRUCTURE subpopulations (
Figure 5). Gaur and Tho-tho cattle grouped close to each other in the same group-I, similar to model-based subpopulation 1. However, gaur and Tho-tho have distinct subgroups within group-I. This result is similar to the neighbor-joining clustering pattern and further supported the results of the neighbor-joining clustering, and indicated that gaur and Tho-tho are different from each other. The PCoA results for Nagaland and Arunachal mithun, along with a few Mizoram mithun grouped in top right-side group-II, are similar to the model-based STRUCTURE analysis. Similarly, Manipur and Mizoram mithun grouped in the top left-side of group-III, similar to the model-based SP3 subpopulation grouping.
The outcome of the distance-based neighbor-joining tree, PCoA, and PCA analysis were highly congruent with the results of the model-based STRUCTURE analysis, which indicated the accurate grouping of mithun, gaur, and Tho-tho genotypes and further supported the results of STRUCTURE. These analyses revealed that mithun, gaur, and Tho-tho cattle are different populations and distinct from each other.
3.8. Hardy–Weinberg Test
All 19 microsatellite loci showed significant deviation from the Hardy–Weinberg equilibrium (
p < 0.05) in Indian mithun and gaur. Estimates of within-population inbreeding coefficient (F
IS) as per [
53] were 30.00% and 6.50% in the mithun and gaur population, respectively, which was high and positive (
p < 0.05).
3.9. Bottleneck Effect
Our bottleneck analysis exhibited significant heterozygosity excess under IAM model in the mithun, gaur, and Tho-tho cattle population (p < 0.01), while heterozygosity excess was detected only in the gaur population (p < 0.05) in the TPM model. No heterozygosity excess was found in any population in the SMM model. This revealed a minor genetic bottleneck in recent history only in the gaur population.
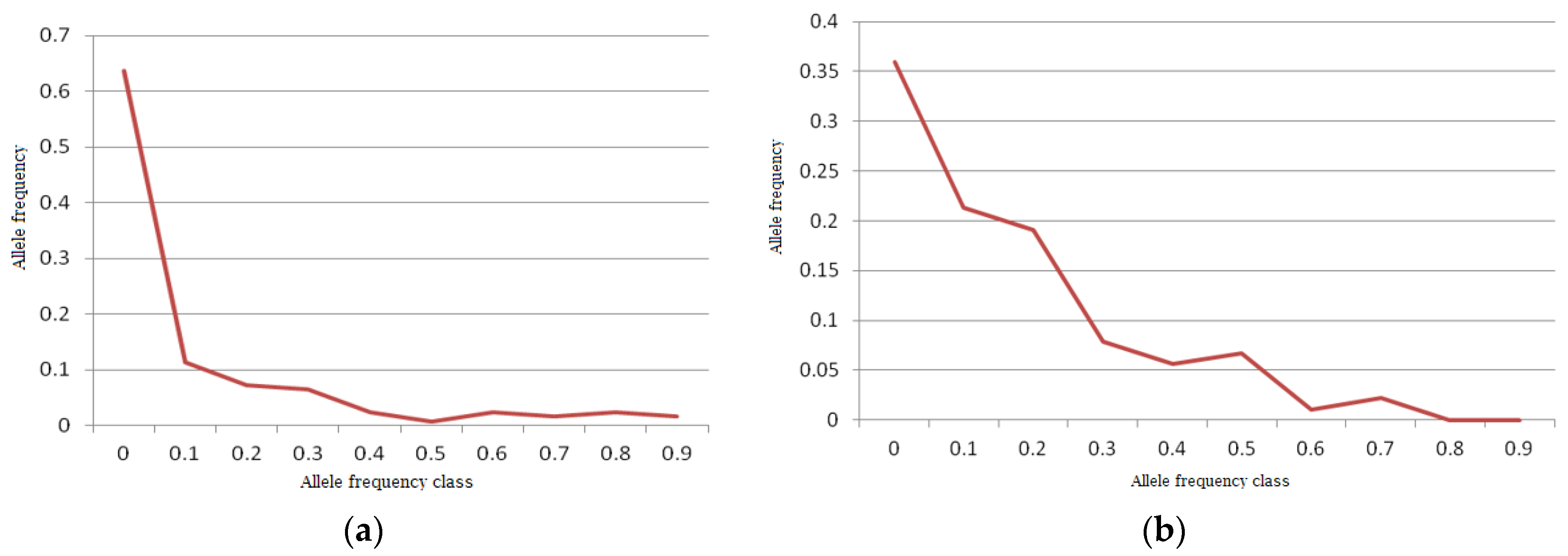
A normal L-shaped distribution of allelic frequencies was found in the bottleneck analysis in the Indian mithun population by the qualitative graphical test (
Figure 6a). There was no mode-shift found in the distribution of allelic frequencies, showing the absence of any genetic bottleneck in the Indian mithun population, similar to Yunnan mithun [
54]. However, the gaur population was found to have shifted mode, indicating the presence of a bottleneck in recent history, as the distribution of allelic frequency did not form an exact L-shaped distribution (
Figure 6b).
3.10. Mitochondrial Genetic Variability
A fragment of 1021 bp transmembrane protein coding CYTB gene of mtDNA was explored resulting in identification of 961 variable sites. We identified 21 haplotypes with an average diversity of 0.62 (
Table 7). An overall estimate for population indices revealed nucleotide diversity of 0.058 and average expected heterozygosity of 0.926. These indices indicated sufficient mtDNA diversity among the analyzed breeds.
The gene diversity examined on the basis of CYTB bp reflected the highest diversity in Mizoram mithun and the lowest in Arunachal mithun as well as gaur. However, the number of polymorphic sites was found to be highest in gaur, while the lowest number was found in Tho-tho cattle. Regarding mithun species only, all the breeds had a similar number of polymorphic sites, except Arunachal mithun, which was very low. The polymorphic sites converged to very few haplotypes, where Manipur mithun was having six haplotypes while gaur had only two haplotypes. The expected heterozygosity was highest in gaur and lowest in Manipur and Nagaland mithun. The highest nucleotide diversity was seen in Gaur, while the lowest was in Tho-tho. With regard to mithun breeds, the highest nucleotide diversity was in Mizoram mithun and the lowest was in Arunachal mithun.
The phylogenetic relationship between different species and the various breeds of mithun was visualized by constructing a phylogenetic tree (
Figure S2) and a cladogram (
Figure S3). Tho-tho cattle was the most distinct, which formed a separate clade than the other considered species in the study. The other ones, mithun and gaur, formed a group which further separated into different clades as expected. Among the species, B. gaurus was genetically closer to Mizoram mithun as compared to other breeds.
5. Conclusions
Our study revealed medium to high within breed genetic diversity in the Indian mithun, gaur, and Tho-tho cattle population, implying moderate genetic differentiation among these bovine species. Absence of any recent genetic bottleneck in the mithun, and Tho-tho and minor genetic bottleneck in the gaur, in spite of very low population size is encouraging for ongoing genetic improvement and conservation efforts.
This study also demonstrated that the cattle microsatellite markers can be used effectively on the mithun and gaur population, which generated valuable information regarding the present genetic status of these rare bovine species. The present level of heterozygosity in the Indian mithuns, gaur, and Tho-tho population was adequate. These findings have helped towards the development and introduction of a rational breeding policy for mithuns in their native tracts and for the wild gaurs for future genetic improvement of these unique bovines and their conservation, which are under threat of extinction.


 ,
, 








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}