
DNA Barcoding and Species Delimitation for Dogfish Sharks Belonging to the Squalus Genus (Squaliformes: Squalidae)

 , ,
, ,  , , , and
, , , and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Barcoding
2.3. Automatic Species Delimitation Analyses
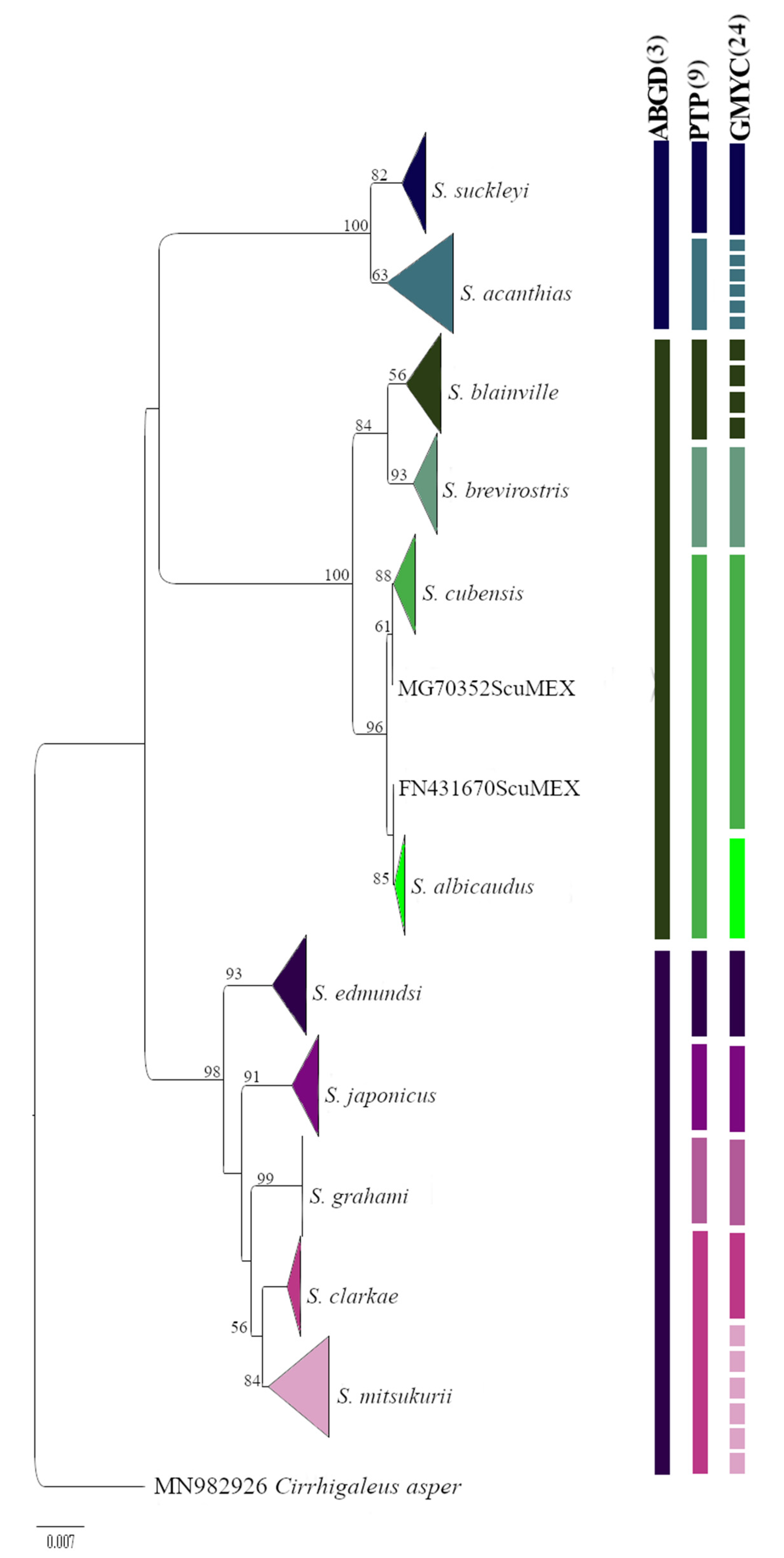
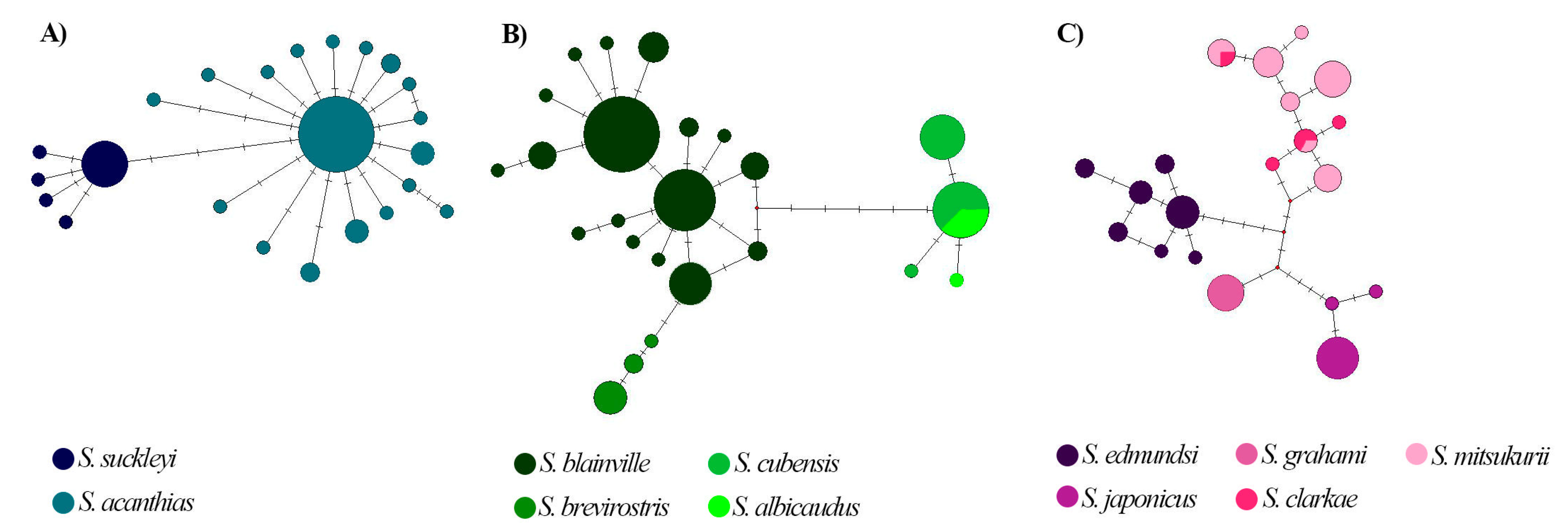
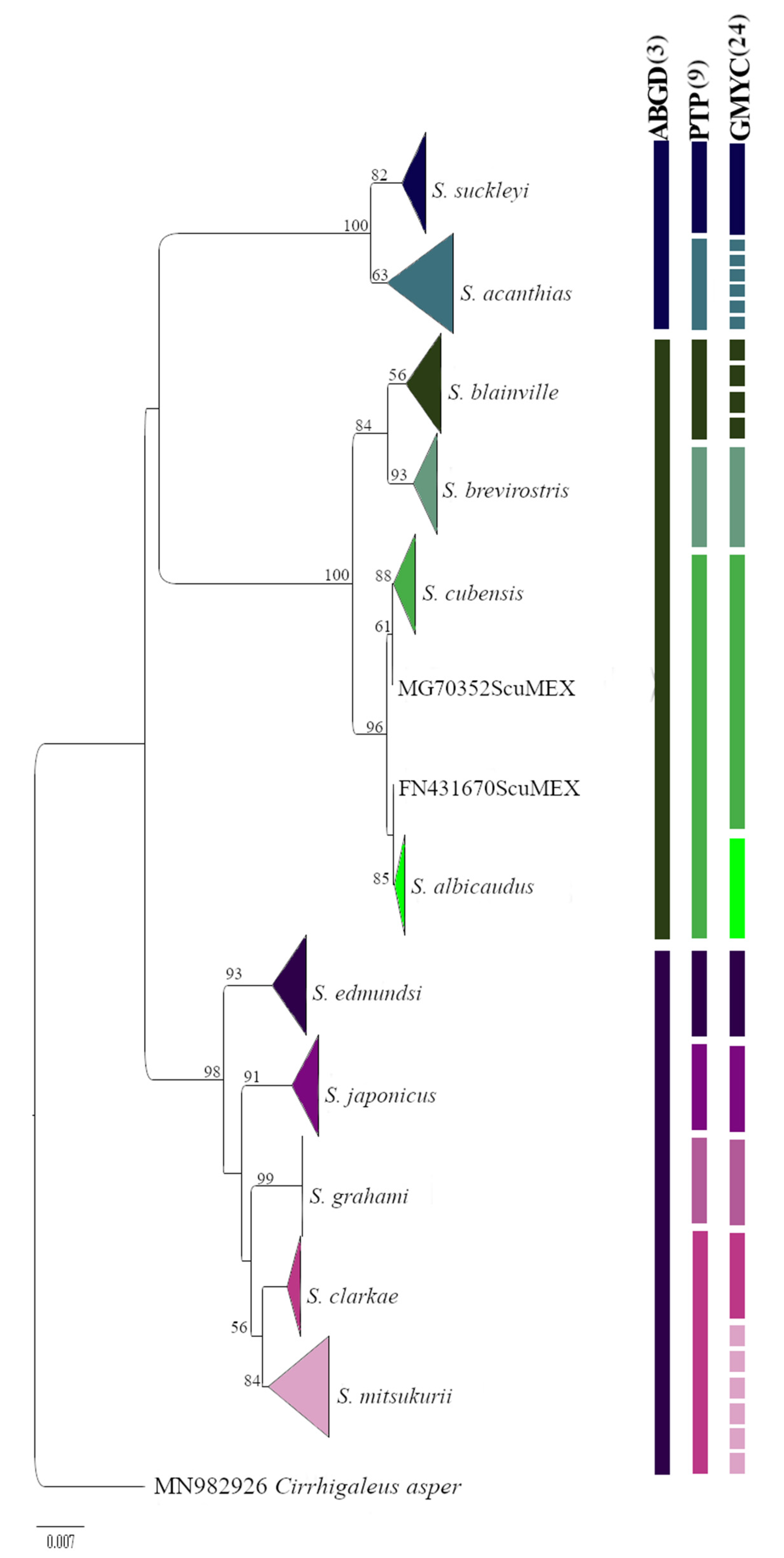
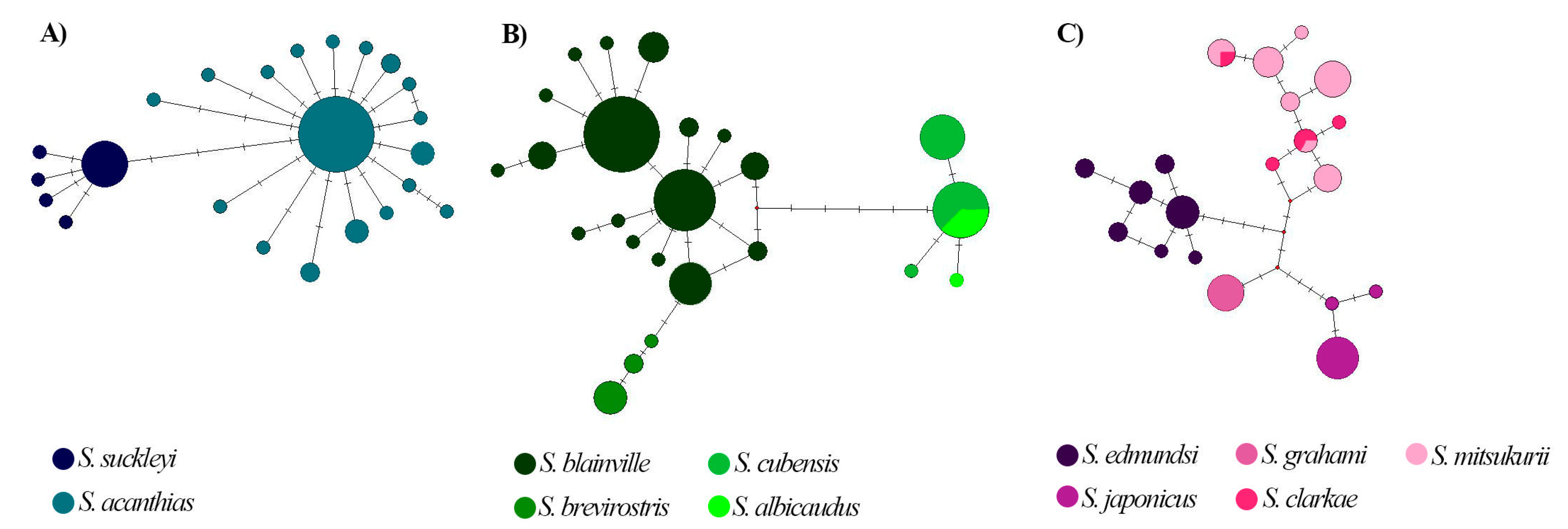
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Last, P.; White, W.; Pogonoski, J. Descriptions of New Dogfishes of the Genus Squalus (Squaloidea: Squalidae); CSIRO Marine and Atmospheric Research: Hobart, Australia, 2007. [Google Scholar]
- Viana, S.T.D.F.; Carvalho, M.R.D.; Gomes, U.L. Taxonomy and Morphology of Species of the Genus Squalus Linnaeus, 1758 from the Southwestern Atlantic Ocean (Chondrichthyes: Squaliformes: Squalidae). Zootaxa 2016, 4133, 1–89. [Google Scholar] [CrossRef] [PubMed]
- Daly-Engel, T.S.; Koch, A.; Anderson, J.M.; Cotton, C.F.; Rubbs, R.D. Description of a New Deep-Water Dogfish Shark from Hawaii, with Comments on the Squalus mitsukurii Species Complex in the West Pacific. Zookeys 2018, 798, 135–157. [Google Scholar] [CrossRef] [PubMed]
- Pfleger, M.O.; Grubbs, R.D.; Cotton, C.F.; Daly-Engel, T.S. Squalus clarkae Sp. Nov., a New Dogfish Shark from the Northwest Atlantic and Gulf of Mexico, with Comments on the Squalus mitsukurii Species Complex. Zootaxa 2018, 4444, 101–119. [Google Scholar] [CrossRef] [PubMed]
- Ebert, D.; Fowler, S.; Compagno, L. Sharks of the World: A Fully Illustrated Guide; Wild Nature Press: Plymouth, MA, USA, 2013; ISBN 978-0-9573946-0-5. [Google Scholar]
- Compagno, L.J. An Annotated and Illustrated Catalogue of Shark Species Known to Date. In FAO Species Catalogue; United Nations Development Programme Food and Agriculture Organization of The United Nations: Rome, Italy, 1984; Volume 4, pp. vii + 249. [Google Scholar]
- Compagno, L.J.V.; Dando, M.; Fowler, S. Sharks of the World; Princeton University Press: Princeton, NJ, USA, 2005. [Google Scholar]
- Cortés, E. Life History Patterns and Correlations in Sharks. Rev. Fish. Sci. 2000, 8, 299–344. [Google Scholar] [CrossRef]
- Ebert, D.; White, W.; Goldman, K.; Compagno, L.; Daly-Engel, T.; Ward, R. Resurrection and Redescription of Squalus suckleyi (Girard, 1854) from the North Pacific, with Comments on the Squalus acanthias Subgroup (Squaliformes: Squalidae). Zootaxa 2010, 2612, 22–40. [Google Scholar] [CrossRef]
- Wourms, J.P. Reproduction and Development in Chondrichthyan Fishes. Am. Zool. 1977, 17, 379–410. [Google Scholar] [CrossRef]
- Dell’Apa, A.; Bangley, C.W.; Rulifson, R.A. Who Let the Dogfish out? A Review of Management and Socio-Economic Aspects of Spiny Dogfish Fisheries. Rev. Fish Biol. Fish. 2015, 25, 273–295. [Google Scholar] [CrossRef]
- Lamarca, F.; Vianna, M.; Vilasboa, A. The First Reproductive Parameters and Evidence of Multiple Paternity in One New Spiny Dogfish Species, Squalus albicaudus (Squaliformes, Squalidae). J. Fish Biol. 2020, 97, 1268–1272. [Google Scholar] [CrossRef]
- Ketchen, K.S. Size at Maturity, Fecundity, and Embryonic Growth of the Spiny Dogfish (Squalus acanthias) in British Columbia Waters. J. Fish. Res. Board Can. 1972, 29, 1717–1723. [Google Scholar] [CrossRef]
- Nammack, M.F.; Musick, J.A.; Colvocoresses, J.A. Life History of Spiny Dogfish off the Northeastern United States. Trans. Am. Fish. Soc. 1985, 114, 367–376. [Google Scholar] [CrossRef]
- Jones, T.S.; Ugland, K.I. Reproduction of Female Spiny Dogfish, Squalus acanthias, in the Oslofjord. Fish. Bull. Natl. Ocean. Atmos. Adm. 2001, 99, 685–690. [Google Scholar]
- Dulvy, N.K.; Fowler, S.L.; Musick, J.A.; Cavanagh, R.D.; Kyne, P.M.; Harrison, L.R.; Carlson, J.K.; Davidson, L.N.; Fordham, S.V.; Francis, M.P.; et al. Extinction Risk and Conservation of the World’s Sharks and Rays. eLife 2014, 3, e00590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- List, I.R.; The IUCN Red List of Threatened Species. Version 2020-1. 2020. Available online: https://www.iucnredlist.org (accessed on 21 March 2021).
- MMA; Bataus, Y.S.D.L.; Nogueira, C.D.C.; Marcovaldi, M.A.; Vogt, R.C.; Coutinho, M.E.; Werneck, F.D.P.; Martins, M.R.C.; Colli, G.R. Livro Vermelho da Fauna Brasileira Ameaçada de Extinção; ICMBio, MMA Distrito Federal; Instituto Chico Mendes de Conservação da Biodiversidade: Brasília, Brazil, 2018. [Google Scholar]
- Geraci, M.L.; Ragonese, S.; Norrito, G.; Scannella, D.; Falsone, F.; Vitale, S. A Tale on the Demersal and Bottom Dwelling Chondrichthyes in the South of Sicily through 20 Years of Scientific Survey. In Chondrichthyes—Multidisciplinary Approach; IntechOpen: London, UK, 2017; pp. 13–37. [Google Scholar]
- Last, P.R.; White, W.T.; Pogonoski, J.J.; Gledhill, D.C.; Yearsley, G.K.; Ward, R.D. Application of a Rapid Taxonomic Approach to the Genus Squalus; CSIRO Marine and Atmospheric Research: Hobart, Australia, 2007. [Google Scholar]
- Veríssimo, A.; Zaera-Perez, D.; Leslie, R.; Iglésias, S.P.; Séret, B.; Grigoriou, P.; Sterioti, A.; Gubili, C.; Barría, C.; Duffy, C.; et al. Molecular Diversity and Distribution of Eastern Atlantic and Mediterranean Dogfishes Squalus Highlight Taxonomic Issues in the Genus. Zool. Scr. 2017, 46, 414–428. [Google Scholar] [CrossRef]
- Bellodi, A.; Porcu, C.; Cau, A.; Marongiu, M.F.; Melis, R.; Mulas, A.; Pesci, P.; Follesa, M.C.; Cannas, R. Investigation on the Genus Squalus in the Sardinian Waters (Central-Western Mediterranean) with Implications on Its Management. Mediterr. Mar. Sci. 2018, 19, 256–272. [Google Scholar] [CrossRef] [Green Version]
- Díaz-Jaimes, P.; Bayona-Vásquez, N.J.; Escatel-Luna, E.; Uribe-Alcocer, M.; Pecoraro, C.; Adams, D.H.; Frazier, B.S.; Glenn, T.C.; Babbucci, M. Population Genetic Divergence of Bonnethead Sharks Sphyrna tiburo in the Western North Atlantic: Implications for Conservation. Aquat. Conserv. Mar. Freshw. Ecosyst. 2021, 31, 83–98. [Google Scholar] [CrossRef]
- Ceballos, G.; Ehrlich, P.R.; Barnosky, A.D.; García, A.; Pringle, R.M.; Palmer, T.M. Accelerated Modern Human–Induced Species Losses: Entering the Sixth Mass Extinction. Sci. Adv. 2015, 1, e1400253. [Google Scholar] [CrossRef] [Green Version]
- Ziadi-Künzli, F.; Soliman, T.; Imai, H.; Sakurai, M.; Maeda, K.; Tachihara, K. Re-Evaluation of Deep-Sea Dogfishes (Genus Squalus) in Japan Using Phylogenetic Inference. Deep. Sea Res. Part I Oceanogr. Res. Pap. 2020, 160, 103261. [Google Scholar] [CrossRef]
- Ferrari, A.; Di Crescenzo, S.; Cariani, A.; Crobe, V.; Benvenuto, A.; Piattoni, F.; Mancusi, C.; Bonnici, L.; Bonello, J.J.; Schembri, P.J.; et al. Puzzling over Spurdogs: Molecular Taxonomy Assessment of the Squalus Species in the Strait of Sicily. Eur. Zool. J. 2021, 88, 181–190. [Google Scholar] [CrossRef]
- Naylor, G.J.P.; Caira, J.N.; Jensen, K.; Rosana, K.A.M.; White, W.T.; Last, P.R. A DNA Sequence-Based Approach to the Identification of Shark and Ray Species and Its Implications for Global Elasmobranch Diversity and Parasitology; Bulletin of the American Museum of Natural History, No. 367; DNA Identification of Sharks and Rays; American Museum of Natural History—Scientific Publications: New York, NY, USA, 2012. [Google Scholar]
- Vella, A.; Vella, N.; Schembri, S. A Molecular Approach towards Taxonomic Identification of Elasmobranch Species from Maltese Fisheries Landings. Mar. Genom. 2017, 36, 17–23. [Google Scholar] [CrossRef]
- Ward, R.D.; Holmes, B.H.; Zemlak, T.S.; Smith, P.J. Part 12—DNA Barcoding Discriminates Spurdogs of the Genus Squalus. In Descriptions of New Dogfishes of the Genus Squalus (Squaloidea: Squalidae); CSIRO Marine and Atmospheric Research: Hobart, Australia, 2007; pp. 117–130. [Google Scholar]
- Ward, R.D. DNA Barcode Divergence among Species and Genera of Birds and Fishes. Mol. Ecol. Resour. 2009, 9, 1077–1085. [Google Scholar] [CrossRef]
- Mabragaña, E.; Díaz de Astarloa, J.M.; Hanner, R.; Zhang, J.; González Castro, M. DNA Barcoding Identifies Argentine Fishes from Marine and Brackish Waters. PLoS ONE 2011, 6, e28655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hara, Y.; Yamaguchi, K.; Onimaru, K.; Kadota, M.; Koyanagi, M.; Keeley, S.D.; Tatsumi, K.; Tanaka, K.; Motone, F.; Kageyama, Y. Shark Genomes Provide Insights into Elasmobranch Evolution and the Origin of Vertebrates. Nat. Ecol. Evol. 2018, 2, 1761–1771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ovenden, J.R.; Dudgeon, C.; Feutry, P.; Feldheim, K.; Maes, G.E. Genetics and Genomics for Fundamental and Applied Research on Elasmobranchs. In Shark Research: Emerging Technologies and Applications for the Field and Laboratory; CRC Press: Boca Raton, FL, USA, 2019; pp. 235–253. [Google Scholar]
- Pazmiño, D.A.; van Herderden, L.; Simpfendorfer, C.A.; Junge, C.; Donnellan, S.C.; Hoyos-Padilla, E.M.; Duffy, C.A.; Huveneers, C.; Gillanders, B.M.; Butcher, P.A. Introgressive Hybridisation between Two Widespread Sharks in the East Pacific Region. Mol. Phylogenet. Evol. 2019, 136, 119–127. [Google Scholar] [CrossRef]
- Delaval, A.; Frost, M.; Bendall, V.; Hetherington, S.J.; Stirling, D.; Hoarau, G.; Jones, C.S.; Noble, L.R. Population and Seascape Genomics of a Critically Endangered Benthic Elasmobranch, the Blue Skate Dipturus batis. Evol. Appl. 2022, 15, 78–94. [Google Scholar] [CrossRef] [PubMed]
- Henderson, A.C.; Reeve, A.J.; Jabado, R.W.; Naylor, G.J. Taxonomic Assessment of Sharks, Rays and Guitarfishes (Chondrichthyes: Elasmobranchii) from South-Eastern Arabia, Using the NADH Dehydrogenase Subunit 2 (NADH2) Gene. Zool. J. Linn. Soc. 2016, 176, 399–442. [Google Scholar] [CrossRef] [Green Version]
- Ward, R.D.; Holmes, B.H.; White, W.T.; Last, P.R. DNA Barcoding Australasian Chondrichthyans: Results and Potential Uses in Conservation. Mar. Freshw. Res. 2008, 59, 57–71. [Google Scholar] [CrossRef]
- Hellberg, R.S.; Isaacs, R.B.; Hernandez, E.L. Identification of Shark Species in Commercial Products Using DNA Barcoding. Fish. Res. 2019, 210, 81–88. [Google Scholar] [CrossRef]
- Puillandre, N.; Lambert, A.; Brouillet, S.; Achaz, G. ABGD, Automatic Barcode Gap Discovery for Primary Species Delimitation. Mol. Ecol. 2012, 21, 1864–1877. [Google Scholar] [CrossRef]
- Zhang, J.; Kapli, P.; Pavlidis, P.; Stamatakis, A. A General Species Delimitation Method with Applications to Phylogenetic Placements. Bioinformatics 2013, 29, 2869–2876. [Google Scholar] [CrossRef] [Green Version]
- Pons, J.; Barraclough, T.G.; Gomez-Zurita, J.; Cardoso, A.; Duran, D.P.; Hazell, S.; Kamoun, S.; Sumlin, W.D.; Vogler, A.P. Sequence-Based Species Delimitation for the DNA Taxonomy of Undescribed Insects. Syst. Biol. 2006, 55, 595–609. [Google Scholar] [CrossRef] [Green Version]
- Fujisawa, T.; Barraclough, T.G. Delimiting Species Using Single-Locus Data and the Generalized Mixed Yule Coalescent Approach: A Revised Method and Evaluation on Simulated Data Sets. Syst. Biol. 2013, 62, 707–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melo, B.F.; Sato, Y.; Foresti, F.; Oliveira, C. The Roles of Marginal Lagoons in the Maintenance of Genetic Diversity in the Brazilian Migratory Fishes Prochilodus argenteus and P. costatus. Neotrop. Ichthyol. 2013, 11, 625–636. [Google Scholar] [CrossRef] [Green Version]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An Integrated and Extendable Desktop Software Platform for the Organization and Analysis of Sequence Data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: A Multiple Sequence Alignment Method with Reduced Time and Space Complexity. BMC Bioinform. 2004, 5, 113. [Google Scholar] [CrossRef] [Green Version]
- Kousteni, V.; Kasapidis, P.; Kotoulas, G.; Megalofonou, P. Evidence of High Genetic Connectivity for the Longnose Spurdog Squalus blainville in the Mediterranean Sea. Mediterr. Mar. Sci. 2016, 17, 371–383. [Google Scholar] [CrossRef]
- Knebelsberger, T.; Landi, M.; Neumann, H.; Kloppmann, M.; Sell, A.F.; Campbell, P.D.; Laakmann, S.; Raupach, M.J.; Carvalho, G.R.; Costa, F.O. A Reliable DNA Barcode Reference Library for the Identification of the North European Shelf Fish Fauna. Mol. Ecol. Resour. 2014, 14, 1060–1071. [Google Scholar] [CrossRef]
- Landi, M.; Dimech, M.; Arculeo, M.; Biondo, G.; Martins, R.; Carneiro, M.; Carvalho, G.R.; Lo Brutto, S.; Costa, F.O. DNA Barcoding for Species Assignment: The Case of Mediterranean Marine Fishes. PLoS ONE 2014, 9, e106135. [Google Scholar] [CrossRef]
- Stelbrink, B.; von Rintelen, T.; Cliff, G.; Kriwet, J. Molecular Systematics and Global Phylogeography of Angel Sharks (Genus Squatina). Mol. Phylogenetics Evol. 2010, 54, 395–404. [Google Scholar] [CrossRef]
- Almerón-Souza, F.; Sperb, C.; Castilho, C.L.; Figueiredo, P.I.C.C.; Gonçalves, L.T.; Machado, R.; Oliveira, L.R.; Valiati, V.H.; Fagundes, N.J.R. Molecular Identification of Shark Meat from Local Markets in Southern Brazil Based on DNA Barcoding: Evidence for Mislabeling and Trade of Endangered Species. Front. Genet. 2018, 9, 138. [Google Scholar] [CrossRef]
- McCusker, M.R.; Denti, D.; Van Guelpen, L.; Kenchington, E.; Bentzen, P. Barcoding Atlantic Canada’s Commonly Encountered Marine Fishes. Mol. Ecol. Resour. 2013, 13, 177–188. [Google Scholar] [CrossRef]
- Steinke, D.; Zemlak, T.S.; Gavin, H.; Hebert, P.D.N. DNA Barcoding Fishes of the Canadian Pacific. Mar. Biol. 2009, 156, 2641–2647. [Google Scholar] [CrossRef]
- Gkafas, G.A.; Megalofonou, P.; Batzakas, G.; Apostolidis, A.P.; Exadactylos, A. Molecular Phylogenetic Convergence within Elasmobranchii Revealed by Cytochrome Oxidase Subunits. Biochem. Syst. Ecol. 2015, 61, 510–515. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Kimura, M. A Simple Method for Estimating Evolutionary Rates of Base Substitutions through Comparative Studies of Nucleotide Sequences. J. Mol. Evol. 1980, 16, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Felsenstein, J. Confidence Limits on Phylogenies: An Approach Using the Bootstrap. Evolution 1985, 39, 783–791. [Google Scholar] [CrossRef] [PubMed]
- Librado, P.; Rozas, J. DnaSP v5: A Software for Comprehensive Analysis of DNA Polymorphism Data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [Green Version]
- Leigh, J.W.; Bryant, D. POPART: Full-feature Software for Haplotype Network Construction. Methods Ecol. Evol. 2015, 6, 1110–1116. [Google Scholar] [CrossRef]
- Ward, R.D.; Zemlak, T.S.; Innes, B.H.; Last, P.R.; Hebert, P.D. DNA Barcoding Australia’s Fish Species. Philos. Trans. R. Soc. B Biol. Sci. 2005, 360, 1847–1857. [Google Scholar] [CrossRef]
- Hubert, N.; Hanner, R.; Holm, E.; Mandrak, N.E.; Taylor, E.; Burridge, M.; Watkinson, D.; Dumont, P.; Curry, A.; Bentzen, P.; et al. Identifying Canadian Freshwater Fishes through DNA Barcodes. PLoS ONE 2008, 3, e2490. [Google Scholar] [CrossRef] [Green Version]
- Pereira, L.H.; Hanner, R.; Foresti, F.; Oliveira, C. Can DNA Barcoding Accurately Discriminate Megadiverse Neotropical Freshwater Fish Fauna? BMC Genet. 2013, 14, 20. [Google Scholar] [CrossRef] [Green Version]
- Ramirez, J.L.; Birindelli, J.L.; Carvalho, D.C.; Affonso, P.R.A.M.; Venere, P.C.; Ortega, H.; Carrillo-Avila, M.; Rodríguez-Pulido, J.A.; Galetti, P.M. Revealing Hidden Diversity of the Underestimated Neotropical Ichthyofauna: DNA Barcoding in the Recently Described Genus Megaleporinus (Characiformes: Anostomidae). Front. Genet. 2017, 8, 149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramirez, J.L.; Galetti, P.M. DNA Barcode and Evolutionary Relationship within Laemolyta Cope 1872 (Characiformes: Anostomidae) through Molecular Analyses. Mol. Phylogenet. Evol. 2015, 93, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Cerqueira, N.N.C.D.; Rotundo, M.M.; Marceniuk, A.P.; da Cruz, V.P.; Foresti, F.; Oliveira, C. Molecular Identification of Brachygenys and Haemulon Species (Perciformes: Haemulidae) from the Brazilian Coast. Neotrop. Ichthyol. 2021, 19. [Google Scholar] [CrossRef]
- Ortiz, D.; Francke, O.F. Two DNA Barcodes and Morphology for Multi-Method Species Delimitation in Bonnetina tarantulas (Araneae: Theraphosidae). Mol. Phylogenet. Evol. 2016, 101, 176–193. [Google Scholar] [CrossRef] [PubMed]
- Carstens, B.C.; Pelletier, T.A.; Reid, N.M.; Satler, J.D. How to Fail at Species Delimitation. Mol. Ecol. 2013, 22, 4369–4383. [Google Scholar] [CrossRef] [PubMed]
- Borsa, P.; Shen, K.-N.; Arlyza, I.S.; Hoareau, T.B. Multiple Cryptic Species in the Blue-Spotted Maskray (Myliobatoidei: Dasyatidae: Neotrygon Spp.): An Update. C. R. Biol. 2016, 339, 417–426. [Google Scholar] [CrossRef]
- Borsa, P.; Arlyza, I.S.; Hoareau, T.B.; Shen, K.-N. Diagnostic Description and Geographic Distribution of Four New Cryptic Species of the Blue-Spotted Maskray Species Complex (Myliobatoidei: Dasyatidae; Neotrygon Spp.) Based on DNA Sequences. J. Ocean. Limnol. 2018, 36, 827–841. [Google Scholar] [CrossRef] [Green Version]
- Bonello, J.J.; Bonnici, L.; Ferrari, A.; Cariani, A.; Schembri, P.J. Not All That Clear Cut: Intraspecific Morphological Variability in Squalus blainville (Risso, 1827) and Implications for Identification of the Species. J. Mar. Biol. Assoc. U. K. 2016, 96, 1585–1596. [Google Scholar] [CrossRef]
- Viana, S.T.F.; de Carvalho, M.R.; Ebert, D.A. Squalus bassi Sp. Nov., a New Long-Snouted Spurdog (Chondrichthyes: Squaliformes: Squalidae) from the Agulhas Bank. J. Fish Biol. 2017, 91, 1178–1207. [Google Scholar] [CrossRef]
- Viana, S.T.; de Carvalho, M.R. Squalus shiraii Sp. Nov. (Squaliformes, Squalidae), a New Species of Dogfish Shark from Japan with Regional Nominal Species Revisited. Zoosyst. Evol. 2020, 96, 275. [Google Scholar] [CrossRef]
- Schlick-Steiner, B.C.; Steiner, F.M.; Seifert, B.; Stauffer, C.; Christian, E.; Crozier, R.H. Integrative Taxonomy: A Multisource Approach to Exploring Biodiversity. Annu. Rev. Entomol. 2010, 55, 421–438. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.C.; White, W.T.; Then, A.Y.; Naylor, G.J.; Arunrugstichai, S.; Loh, K.-H. Integrated Taxonomy Revealed Genetic Differences in Morphologically Similar and Non-Sympatric Scoliodon macrorhynchos and S. laticaudus. Animals 2022, 12, 681. [Google Scholar] [CrossRef] [PubMed]
- Petean, F.F.; Naylor, G.J.; Lima, S.M. Integrative Taxonomy Identifies a New Stingray Species of the Genus Hypanus rafinesque, 1818 (Dasyatidae, Myliobatiformes), from the Tropical Southwestern Atlantic. J. Fish Biol. 2020, 97, 1120–1142. [Google Scholar] [CrossRef] [PubMed]
- Cerutti-Pereyra, F.; Meekan, M.G.; Wei, N.-W.V.; O’Shea, O.; Bradshaw, C.J.A.; Austin, C.M. Identification of Rays through DNA Barcoding: An Application for Ecologists. PLoS ONE 2012, 7, e36479. [Google Scholar] [CrossRef] [Green Version]
- Gabbanelli, V.; Díaz de Astarloa, J.M.; Gonzalez-Castro, M.; Vazquez, D.M.; Mabragaña, E. Almost a Century of Oblivion: Integrative Taxonomy Allows the Resurrection of the Longnose Skate Zearaja brevicaudata (Marini, 1933) (Rajiformes; Rajidae). C. R. Biol. 2018, 341, 454–470. [Google Scholar] [CrossRef]
- García-Melo, J.E.; Oliveira, C.; Silva, G.J.D.C.; Ochoa-Orrego, L.E.; Pereira, L.H.G.; Maldonado-Ocampo, J.A. Species Delimitation of Neotropical Characins (Stevardiinae): Implications for Taxonomy of Complex Groups. PLoS ONE 2019, 14, e0216786. [Google Scholar] [CrossRef]
- Meiklejohn, K.A.; Damaso, N.; Robertson, J.M. Assessment of BOLD and GenBank—Their Accuracy and Reliability for the Identification of Biological Materials. PLoS ONE 2019, 14, e0217084. [Google Scholar] [CrossRef] [Green Version]
- Pentinsaari, M.; Ratnasingham, S.; Miller, S.E.; Hebert, P.D.N. BOLD and GenBank Revisited—Do Identification Errors Arise in the Lab or in the Sequence Libraries? PLoS ONE 2020, 15, e0231814. [Google Scholar] [CrossRef]
- Ip, Y.C.A.; Chang, J.J.M.; Lim, K.K.P.; Jaafar, Z.; Wainwright, B.J.; Huang, D. Seeing through Sedimented Waters: Environmental DNA Reduces the Phantom Diversity of Sharks and Rays in Turbid Marine Habitats. BMC Ecol. Evol. 2021, 21, 166. [Google Scholar] [CrossRef]
- Feitosa, L.M.; Martins, A.P.B.; Giarrizzo, T.; Macedo, W.; Monteiro, I.L.; Gemaque, R.; Nunes, J.L.S.; Gomes, F.; Schneider, H.; Sampaio, I. DNA-Based Identification Reveals Illegal Trade of Threatened Shark Species in a Global Elasmobranch Conservation Hotspot. Sci. Rep. 2018, 8, 3347. [Google Scholar] [CrossRef]
- O’Bryhim, J.R.; Parsons, E.C.M.; Lance, S.L. Forensic Species Identification of Elasmobranchs Landed in Costa Rican Artisanal Fisheries. Fish. Res. 2021, 233, 105755. [Google Scholar] [CrossRef]
- Bellodi, A.; Benvenuto, A.; Melis, R.; Mulas, A.; Barone, M.; Barría, C.; Cariani, A.; Carugati, L.; Chatzispyrou, A.; Desrochers, M.; et al. Call Me by My Name: Unravelling the Taxonomy of the Gulper Shark Genus Centrophorus in the Mediterranean Sea through an Integrated Taxonomic Approach. Zool. J. Linn. Soc. 2022, 195, 815–840. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Species | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1—S. suckleyi | 0.0008 | 0.0031 | 0.0118 | 0.0119 | 0.0108 | 0.0109 | 0.0099 | 0.0105 | 0.0101 | 0.0095 | 0.0097 |
| 2—S. acanthias | 0.0077 | 0.0022 | 0.0117 | 0.0123 | 0.0110 | 0.0107 | 0.0099 | 0.0112 | 0.0107 | 0.0101 | 0.0101 |
| 3—S. blainville | 0.0785 | 0.0788 | 0.0032 | 0.0039 | 0.0050 | 0.0052 | 0.0105 | 0.0109 | 0.0104 | 0.0105 | 0.0106 |
| 4—S. brevirostris | 0.0792 | 0.0832 | 0.0116 | 0.0018 | 0.0053 | 0.0054 | 0.0106 | 0.0109 | 0.0106 | 0.0106 | 0.0107 |
| 5—S. cubensis | 0.0701 | 0.0748 | 0.0168 | 0.0188 | 0.0013 | 0.0026 | 0.0099 | 0.0103 | 0.0100 | 0.0096 | 0.0100 |
| 6—S. albicaudus | 0.0707 | 0.0729 | 0.0178 | 0.0197 | 0.0072 | 0.0016 | 0.0100 | 0.0102 | 0.0101 | 0.0102 | 0.0097 |
| 7—S. edmundsi | 0.0630 | 0.0636 | 0.0638 | 0.0667 | 0.0614 | 0.0618 | 0.0021 | 0.0054 | 0.0057 | 0.0047 | 0.0049 |
| 8—S. japonicus | 0.0646 | 0.0720 | 0.0655 | 0.0663 | 0.0601 | 0.0600 | 0.0190 | 0.0009 | 0.0051 | 0.0051 | 0.0055 |
| 9—S. grahami | 0.0631 | 0.0703 | 0.0634 | 0.0649 | 0.0603 | 0.0610 | 0.0200 | 0.0169 | 0.0000 | 0.0045 | 0.0048 |
| 10—S. clarkae | 0.0589 | 0.0675 | 0.0636 | 0.0659 | 0.0577 | 0.0661 | 0.0169 | 0.0185 | 0.0133 | 0.0029 | 0.0026 |
| 11—S. mitsukurii | 0.0605 | 0.0659 | 0.0644 | 0.0665 | 0.0638 | 0.0613 | 0.0190 | 0.0213 | 0.0154 | 0.0084 | 0.0043 |
| Species | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 |
|---|---|---|---|---|---|---|---|---|---|
| 1—S. suckleyi | 0.0008 | 0.0031 | 0.0121 | 0.0119 | 0.0108 | 0.0106 | 0.0108 | 0.0104 | 0.0100 |
| 2—S. acanthias | 0.0078 | 0.0022 | 0.0121 | 0.0122 | 0.0107 | 0.0104 | 0.0114 | 0.0110 | 0.0103 |
| 3—S. blainville | 0.0785 | 0.0788 | 0.0038 | 0.0041 | 0.0052 | 0.0107 | 0.0112 | 0.0107 | 0.0106 |
| 4—S. brevirostris | 0.0792 | 0.0832 | 0.0032 | 0.0018 | 0.0056 | 0.0110 | 0.0112 | 0.0108 | 0.0107 |
| 5—S. cubensis +S. albicaudus | 0.0705 | 0.0734 | 0.0176 | 0.0194 | 0.0038 | 0.0100 | 0.0101 | 0.0100 | 0.0096 |
| 6—S. edmundsi | 0.0630 | 0.0636 | 0.0638 | 0.0667 | 0.0617 | 0.0021 | 0.0053 | 0.0055 | 0.0050 |
| 7—S. japonicus | 0.0646 | 0.0720 | 0.0655 | 0.0663 | 0.0600 | 0.0190 | 0.0009 | 0.0051 | 0.0055 |
| 8—S. grahami | 0.0631 | 0.0703 | 0.0634 | 0.0649 | 0.0608 | 0.0200 | 0.0169 | 0.0000 | 0.0045 |
| 9—S. clarkae + S. mitsukurii | 0.0602 | 0.0662 | 0.0642 | 0.0664 | 0.0623 | 0.0186 | 0.0208 | 0.0151 | 0.0055 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ariza, A.A.; Adachi, A.M.C.L.; Roque, P.; Hazin, F.H.V.; Vianna, M.; Rotundo, M.M.; Delpiani, S.M.; de Astarloa, J.M.D.; Delpiani, G.; Oliveira, C.; et al. DNA Barcoding and Species Delimitation for Dogfish Sharks Belonging to the Squalus Genus (Squaliformes: Squalidae). Diversity 2022, 14, 544. https://doi.org/10.3390/d14070544
Ariza AA, Adachi AMCL, Roque P, Hazin FHV, Vianna M, Rotundo MM, Delpiani SM, de Astarloa JMD, Delpiani G, Oliveira C, et al. DNA Barcoding and Species Delimitation for Dogfish Sharks Belonging to the Squalus Genus (Squaliformes: Squalidae). Diversity. 2022; 14(7):544. https://doi.org/10.3390/d14070544
Chicago/Turabian StyleAriza, Ailton A., Aisni M. C. L. Adachi, Pollyana Roque, Fabio H. V. Hazin, Marcelo Vianna, Matheus M. Rotundo, Sergio M. Delpiani, Juan M. Díaz de Astarloa, Gabriela Delpiani, Claudio Oliveira, and et al. 2022. "DNA Barcoding and Species Delimitation for Dogfish Sharks Belonging to the Squalus Genus (Squaliformes: Squalidae)" Diversity 14, no. 7: 544. https://doi.org/10.3390/d14070544
APA StyleAriza, A. A., Adachi, A. M. C. L., Roque, P., Hazin, F. H. V., Vianna, M., Rotundo, M. M., Delpiani, S. M., de Astarloa, J. M. D., Delpiani, G., Oliveira, C., Foresti, F., & Cruz, V. P. (2022). DNA Barcoding and Species Delimitation for Dogfish Sharks Belonging to the Squalus Genus (Squaliformes: Squalidae). Diversity, 14(7), 544. https://doi.org/10.3390/d14070544