
Postglacial Expansion Routes and Mitochondrial Genetic Diversification of the Freshwater Pearl Mussel in Europe and North America

 , ,
, ,  , , and
, , and
Abstract
1. Introduction
2. Materials and Methods
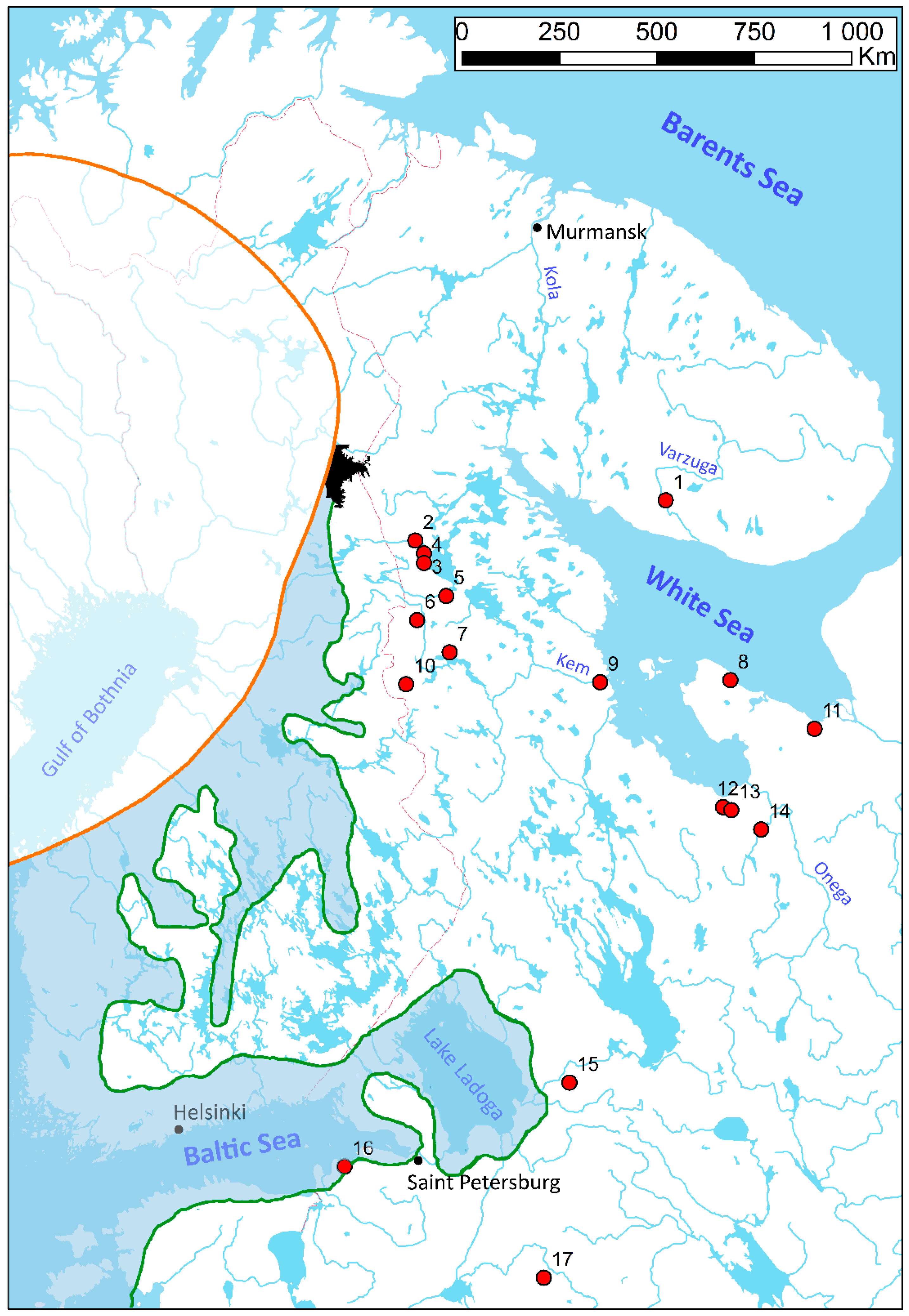
2.1. Sample Collection, DNA Extraction, PCR, and Sequencing
2.2. Genetic Diversity, Genetic Differentiation, and Demographic History
3. Results
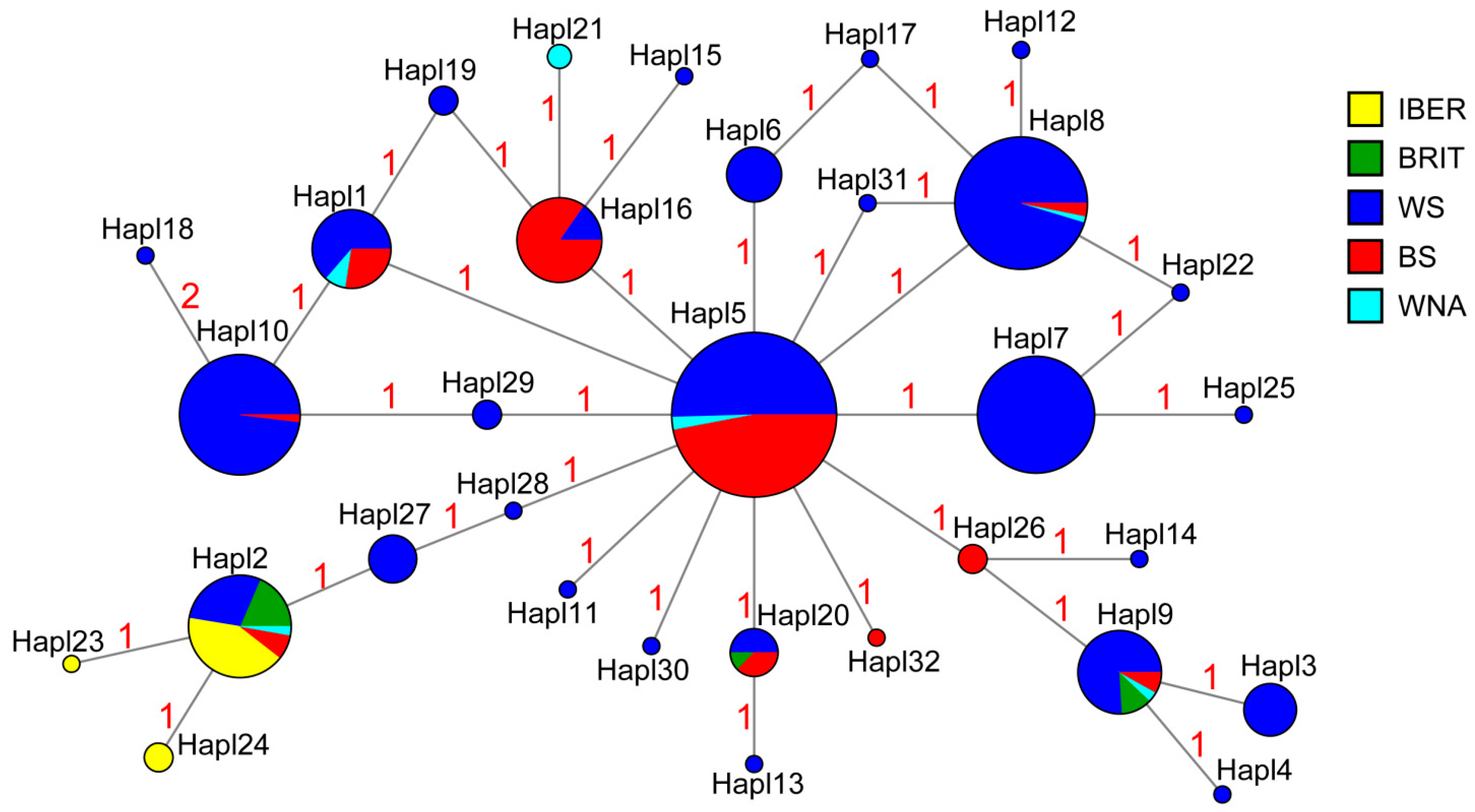
3.1. Genetic Diversity
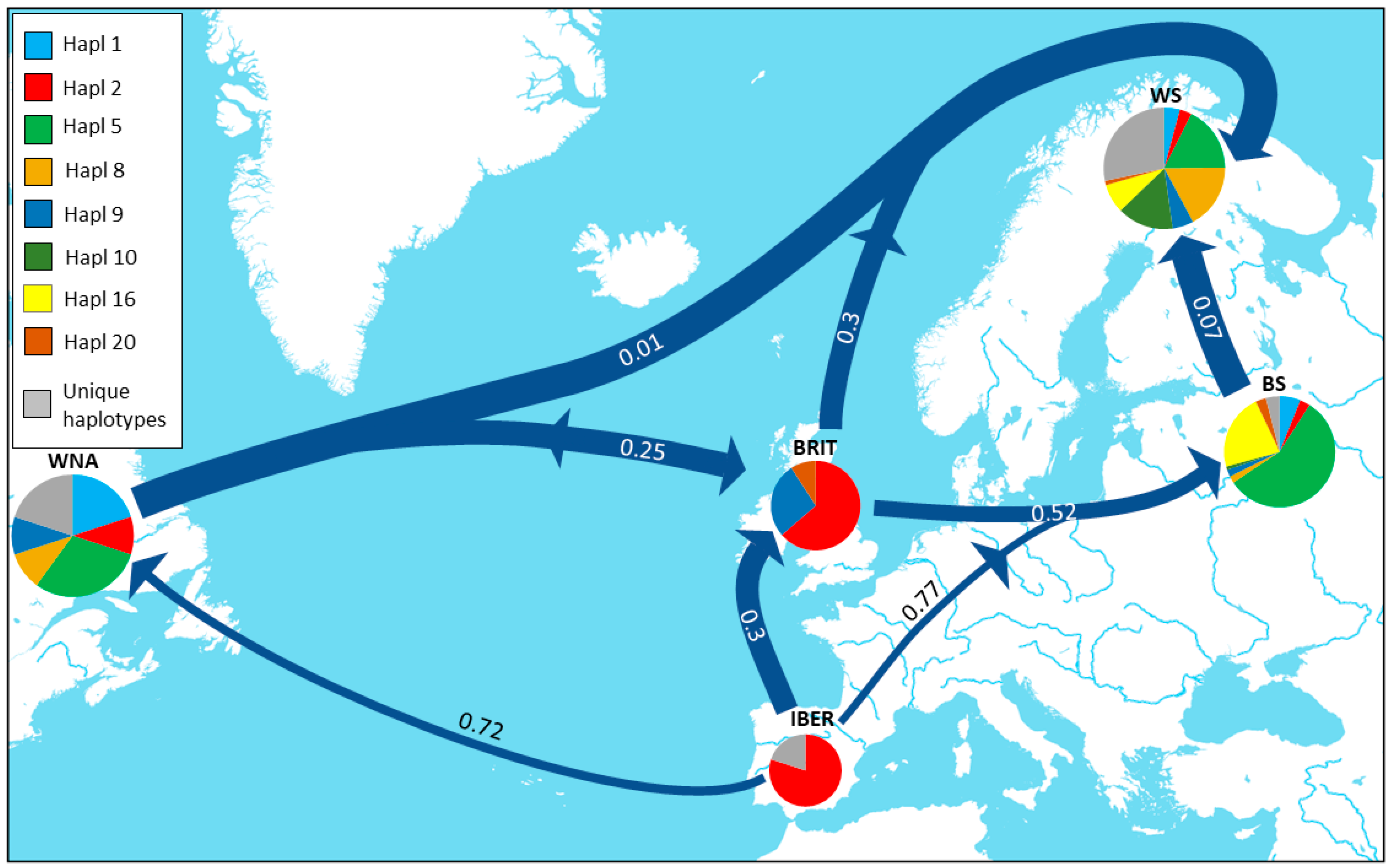
3.2. Genetic Differentiation and Genetic Structure
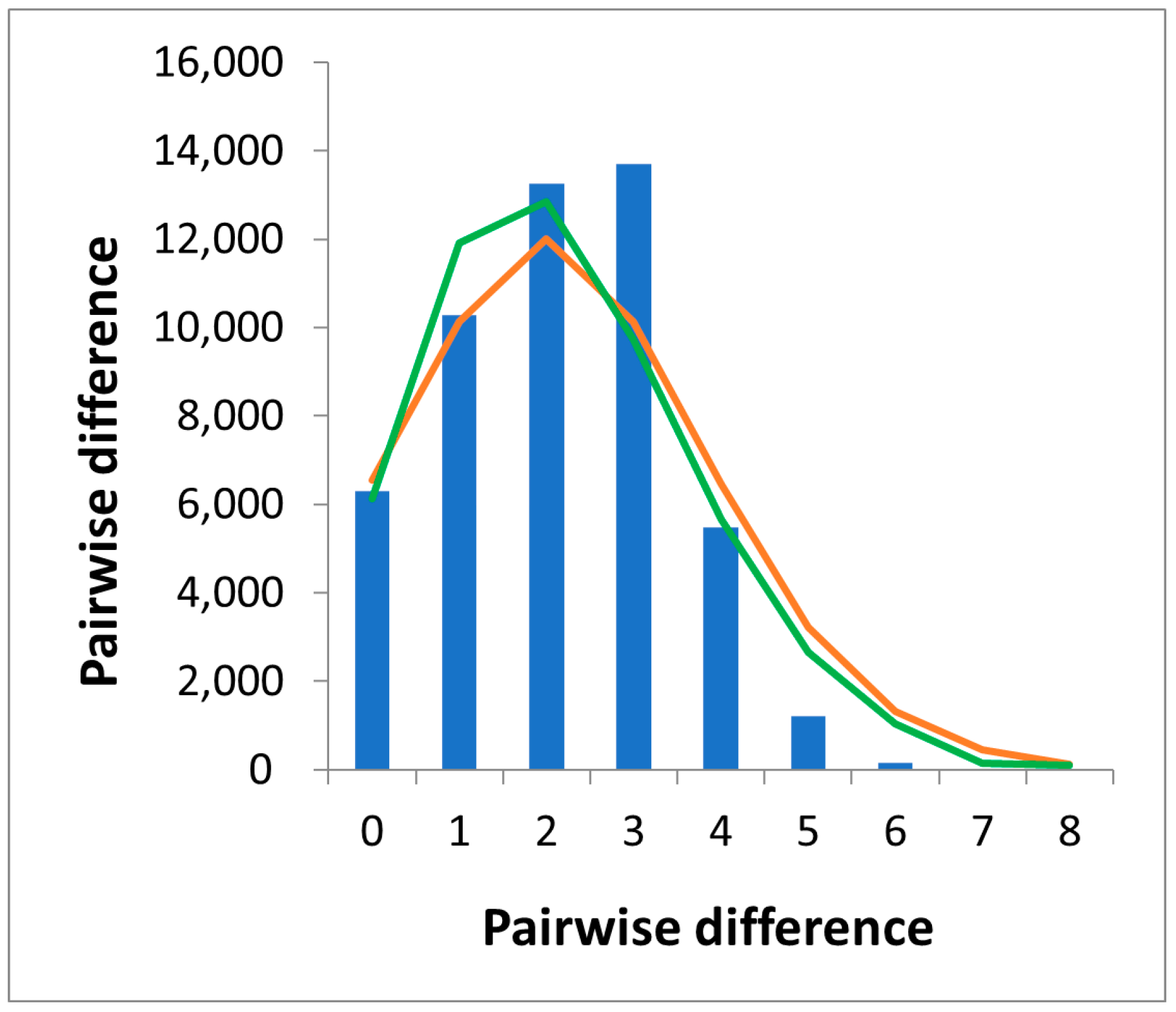
3.3. Demographic Trends
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Geist, J. Strategies for the Conservation of Endangered Freshwater Pearl Mussels (Margaritifera margaritifera L.): A Synthesis of Conservation Genetics and Ecology. Hydrobiologia 2010, 644, 69–88. [Google Scholar] [CrossRef]
- Moorkens, E.A. Addressing the Conservation and Rehabilitation of Margaritifera margaritifera (L.) Populations in the Republic of Ireland within the Framework of the Habitats and Species Directive. J. Conchol. 2010, 40, 339–350. [Google Scholar]
- Makhrov, A.; Bespalaya, J.; Bolotov, I.; Vikhrev, I.; Gofarov, M.; Alekseeva, Y.; Zotin, A. Historical Geography of Pearl Harvesting and Current Status of Populations of Freshwater Pearl Mussel Margaritifera margaritifera (L.) in the Western Part of Northern European Russia. Hydrobiologia 2014, 735, 149–159. [Google Scholar] [CrossRef]
- Bolotov, I.N.; Makhrov, A.A.; Gofarov, M.Y.; Aksenova, O.V.; Aspholm, P.E.; Bespalaya, Y.V.; Kabakov, M.B.; Kolosova, Y.S.; Kondakov, A.V.; Ofenböck, T.; et al. Climate Warming as a Possible Trigger of Keystone Mussel Population Decline in Oligotrophic Rivers at the Continental Scale. Sci. Rep. 2018, 8, 35. [Google Scholar] [CrossRef] [PubMed]
- Young, M.; Williams, J. The Status and Conservation of the Freshwater Pearl Mussel Margaritifera margaritifera Linn. in Great Britain. Biol. Conserv. 1983, 25, 35–52. [Google Scholar] [CrossRef]
- Young, M.R.; Cosgrove, P.J.; Hastie, L. The Extent of, and Causes for, the Decline of a Highly Threatened Naiad: Margaritifera margaritifera. In Cology and Evolutionary Biology of the Freshwater Mussels Unionoidea; Bauer, G., Wachtler, K., Eds.; Springer: Berlin, Germany, 2001; p. 337357. [Google Scholar]
- Karlsson, S.; Larsen, B.M.; Larsen, M.; Hindar, K. Host-Dependent Genetic Variation in Freshwater Pearl Mussel (Margaritifera margaritifera L.). Hydrobiologia 2014, 735, 179–190. [Google Scholar] [CrossRef]
- Geist, J.; Moorkens, E.; Killeen, I.; Feind, S.; Stoeckle, B.C.; Connor, Á.O.; Kuehn, R. Genetic Structure of Irish Freshwater Pearl Mussels (Margaritifera margaritifera and Margaritifera durrovensis): Validity of Subspecies, Roles of Host Fish, and Conservation Implications. Aquat. Conserv. Mar. Freshw. Ecosyst. 2018, 28, 923–933. [Google Scholar] [CrossRef]
- Wacker, S.; Larsen, B.M.; Jakobsen, P.; Karlsson, S. High Levels of Multiple Paternity in a Spermcast Mating Freshwater Mussel. Int. J. Bus. Innov. Res. 2018, 17, 8126–8134. [Google Scholar] [CrossRef]
- Österling, M.; Lopes-Lima, M.; Froufe, E.; Hadzihalilovic, A.H.; Arvidsson, B. The Genetic Diversity and Differentiation of Mussels with Complex Life Cycles and Relations to Host Fish Migratory Traits and Densities. Sci. Rep. 2020, 10, 17435. [Google Scholar] [CrossRef]
- Geist, J.; Söderberg, H.; Karlberg, A.; Kuehn, R. Drainage-Independent Genetic Structure and High Genetic Diversity of Endangered Freshwater Pearl Mussels (Margaritifera margaritifera) in Northern Europe. Conserv. Genet. 2010, 11, 1339–1350. [Google Scholar] [CrossRef]
- Geist, J.; Kuehn, R. Host-Parasite Interactions in Oligotrophic Stream Ecosystems: The Roles of Life-History Strategy and Ecological Niche. Mol. Ecol. 2008, 17, 997–1008. [Google Scholar] [CrossRef] [PubMed]
- Zanatta, D.T.; Stoeckle, B.C.; Inoue, K.; Paquet, A.; Martel, A.L.; Kuehn, R.; Geist, J. High Genetic Diversity and Low Differentiation in North American Margaritifera margaritifera (Bivalvia: Unionida: Margaritiferidae). Biol. J. Linn. Soc. 2018, 123, 850–863. [Google Scholar] [CrossRef]
- Farrington, S.J.; King, R.W.; Baker, J.A.; Gibbons, J.G. Population Genetics of Freshwater Pearl Mussel (Margaritifera margaritifera) in Central Massachusetts and Implications for Conservation. Aquat. Conserv. Mar. Freshw. Ecosyst. 2020, 30, 1945–1958. [Google Scholar] [CrossRef]
- Geist, J.; Kuehn, R. Genetic Diversity and Differentiation of Central European Freshwater Pearl Mussel (Margaritifera margaritifera L.) Populations: Implications for Conservation and Management. Mol. Ecol. 2005, 14, 425–439. [Google Scholar] [CrossRef]
- Bouza, C.; Castro, J.; Martínez, P.; Amaro, R.; Fernández, C.; Ondina, P.; Outeiro, A.; San Miguel, E. Threatened Freshwater Pearl Mussel Margaritifera margaritifera L. in NW Spain: Low and Very Structured Genetic Variation in Southern Peripheral Populations Assessed Using Microsatellite Markers. Conserv. Genet. 2007, 8, 937–948. [Google Scholar] [CrossRef]
- Stoeckle, B.C.; Araujo, R.; Geist, J.; Kuehn, R.; Toledo, C.; Machordom, A. Strong Genetic Differentiation and Low Genetic Diversity of the Freshwater Pearl Mussel (Margaritifera margaritifera L.) in the Southwestern European Distribution Range. Conserv. Genet. 2017, 18, 147–157. [Google Scholar] [CrossRef]
- Taeubert, J.-E.; Geist, J. The Relationship between the Freshwater Pearl Mussel (Margaritifera margaritifera) and Its Hosts. Biol. Bull. 2017, 44, 67–73. [Google Scholar] [CrossRef]
- Bolotov, I.N.; Vikhrev, I.V.; Bespalaya, Y.V.; Gofarov, M.Y.; Kondakov, A.V.; Konopleva, E.S.; Bolotov, N.N.; Lyubas, A.A. Multi-Locus Fossil-Calibrated Phylogeny, Biogeography and a Subgeneric Revision of the Margaritiferidae (Mollusca: Bivalvia: Unionoida). Mol. Phylogenet. Evol. 2016, 103, 104–121. [Google Scholar] [CrossRef]
- Froufe, E.; Sobral, C.; Teixeira, A.; Sousa, R.; Varandas, S.; Aldridge, D.C.; Lopes-Lima, M. Genetic Diversity of the Pan-European Freshwater Mussel Anodonta anatina (Bivalvia: Unionoida) Based on CO1: New Phylogenetic Insights and Implications for Conservation. Aquat. Conserv. Mar. Freshw. Ecosyst. 2014, 24, 561–574. [Google Scholar] [CrossRef]
- Tomilova, A.A.; Lyubas, A.A.; Kondakov, A.V.; Vikhrev, I.V.; Gofarov, M.Y.; Kolosova, Y.S.; Vinarski, M.V.; Palatov, D.M.; Bolotov, I.N. Evidence for Plio-Pleistocene Duck Mussel Refugia in the Azov Sea River Basins. Diversity 2020, 12, 118. [Google Scholar] [CrossRef]
- Consuegra, S.; De Leániz, C.G.; Serdio, A.; González Morales, M.; Straus, L.G.; Knox, D.; Verspoor, E. Mitochondrial DNA Variation in Pleistocene and Modern Atlantic Salmon from the Iberian Glacial Refugium. Mol. Ecol. 2002, 11, 2037–2048. [Google Scholar] [CrossRef] [PubMed]
- Machordom, A.; Araujo, R.; Erpenbeck, D.; Ramos, M.-Á. Phylogeography and Conservation Genetics of Endangered European Margaritiferidae (Bivalvia: Unionoidea). Biol. J. Linn. Soc. 2003, 78, 235–252. [Google Scholar] [CrossRef]
- Välilä, S.J.; Knott, K.E.; Ieshko, E.P.; Veselov, A.E.; Taskinen, J.K. Variation in the COI Gene of the Freshwater Pearl Mussel Margaritifera margaritifera from River Vuokkijoki 1. Biol. Bull. 2017, 44, 92–98. [Google Scholar] [CrossRef]
- Karlsson, S.; Larsen, B.M.; Eriksen, L.; Hagen, M. Four Methods of Nondestructive DNA Sampling from Freshwater Pearl Mussels Margaritifera margaritifera L. (Bivalvia:Unionoida). Freshw. Sci. 2013, 32, 525–530. [Google Scholar] [CrossRef]
- Berg, D.J.; Haag, W.R.; Guttman, S.I.; Sickel, J.B. Mantle Biopsy: A Technique for Nondestructive Tissue-Sampling of Freshwater Mussels. J. N. Am. Benthol. Soc. 1995, 14, 577–581. [Google Scholar] [CrossRef]
- Folmer, O.; Black, M.; Hoeh, W.; Lutz, R.; Vrijenhoek, R. DNA Primers for Amplification of Mitochondrial Cytochrome c Oxidase Subunit I from Diverse Metazoan Invertebrates. Mol. Mar. Biol. Biotechnol. 1994, 3, 294–299. [Google Scholar] [PubMed]
- Bolotov, I.N.; Kondakov, A.V.; Vikhrev, I.V.; Aksenova, O.V.; Bespalaya, Y.V.; Gofarov, M.Y.; Kolosova, Y.S.; Konopleva, E.S.; Spitsyn, V.M.; Tanmuangpak, K.; et al. Ancient River Inference Explains Exceptional Oriental Freshwater Mussel Radiations. Sci. Rep. 2017, 7, 2135. [Google Scholar] [CrossRef]
- Hall, T.A. BioEdit: A User-Friendly Biological Sequence Alignment Editor and Analysis Program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Makhrov, A.A.; Verspoor, E.; Artamonova, V.S.; O’Sullivan, M. Atlantic Salmon Colonization of the Russian Arctic Coast: Pioneers from North America. J. Fish Biol. 2005, 67, 68–79. [Google Scholar] [CrossRef]
- Paradis, E. Pegas: An R Package for Population Genetics with an Integrated–Modular Approach. Bioinformatics 2010, 26, 419–420. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Onsins, S.E.; Rozas, J. Statistical Properties of New Neutrality Tests Against Population Growth. Mol. Biol. Evol. 2002, 19, 2092–2100. [Google Scholar] [CrossRef]
- Excoffier, L.; Lischer, H.E.L. Arlequin Suite Ver 3.5: A New Series of Programs to Perform Population Genetics Analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar] [CrossRef] [PubMed]
- Bandelt, H.J.; Forster, P.; Röhl, A. Median-Joining Networks for Inferring Intraspecific Phylogenies. Mol. Biol. Evol. 1999, 16, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Hammer, D.A.T.; Ryan, P.D.; Hammer, Ø.; Harper, D.A.T. Past: Paleontological Statistics Software Package for Education and Data Analysis. Palaeontol. Electron. 2001, 4, 178. [Google Scholar]
- Stroeven, A.P.; Hättestrand, C.; Kleman, J.; Heyman, J.; Fabel, D.; Fredin, O.; Goodfellow, B.W.; Harbor, J.M.; Jansen, J.D.; Olsen, L.; et al. Deglaciation of Fennoscandia. Quat. Sci. Rev. 2016, 147, 91–121. [Google Scholar] [CrossRef]
- Andrén, T.; Björck, S.; Andrén, E.; Conley, D.; Zillén, L.; Anjar, J. The Development of the Baltic Sea Basin During the Last 130 Ka. In The Baltic Sea Basin; Harff, J., Björck, S., Hoth, P., Eds.; Springer: Berlin/Heidelberg, Germany, 2011; pp. 75–97. ISBN 978-3-642-17219-9. [Google Scholar]
- Johansson, P. The Deglaciation in the Eastern Part of the Weichselian Ice Divide in Finnish Lapland; Geological Snrvey of Finland: Rovaniemi, Finland, 1995. [Google Scholar]
- Hewitt, G.M. Some Genetic Consequences of Ice Ages, and Their Role in Divergence and Speciation. Biol. J. Linn. Soc. 1996, 58, 247–276. [Google Scholar] [CrossRef]
- Bernatchez, L.; Wilson, C.C. Comparative Phylogeography of Nearctic and Palearctic Fishes. Mol. Ecol. 1998, 7, 431–452. [Google Scholar] [CrossRef]
- Berendzen, P.B.; Simons, A.M.; Wood, R.M. Phylogeography of the Northern Hogsucker, Hypentelium Nigricans (Teleostei: Cypriniformes): Genetic Evidence for the Existence of the Ancient Teays River. J. Biogeogr. 2003, 30, 1139–1152. [Google Scholar] [CrossRef]
- Inoue, K.; Monroe, E.M.; Elderkin, C.L.; Berg, D.J. Phylogeographic and Population Genetic Analyses Reveal Pleistocene Isolation Followed by High Gene Flow in a Wide Ranging, but Endangered, Freshwater Mussel. Heredity 2014, 112, 282–290. [Google Scholar] [CrossRef]
- Kimura, M. The Neutral Theory of Molecular Evolution; Cambridge University Press: Cambridge, UK, 1983; ISBN 9780521231091. [Google Scholar]
- Popov, I.Y.; Ostrovsky, A.N. Survival and Extinction of the Southern Populations of Freshwater Pearl Mussel Margaritifera margaritifera in Russia (Leningradskaya and Novgorodskaya Oblast). Hydrobiologia 2014, 735, 161–177. [Google Scholar] [CrossRef]
- Walters, A.D.; Taynor, K.N.; Berg, D.J. Genetic Diversity in the Threatened Freshwater Mussel Lampsilis Powellii. Freshw. Mollusk Biol. Conserv. 2021, 24, 26–33. [Google Scholar] [CrossRef]
- Hague, M.T.J.; Routman, E.J. Does Population Size Affect Genetic Diversity? A Test with Sympatric Lizard Species. Heredity 2016, 116, 92–98. [Google Scholar] [CrossRef]
- Furlan, E.; Stoklosa, J.; Griffiths, J.; Gust, N.; Ellis, R.; Huggins, R.M.; Weeks, A.R. Small Population Size and Extremely Low Levels of Genetic Diversity in Island Populations of the Platypus, Ornithorhynchus anatinus. Ecol. Evol. 2012, 2, 844. [Google Scholar] [CrossRef] [PubMed]
- Bauer, G. Reproductive Strategy of the Freshwater. J. Anim. Ecol. 1987, 56, 691–704. [Google Scholar] [CrossRef]
- Grande, C.; Araujo, R.; Ramos, M.A. The Gonads of Margaritifera auricularia (Spengler, 1793) and M. Margaritifera (Linnaeus, 1758) (Bivalvia: Unionoidea). J. Molluscan Stud. 2001, 67, 27–35. [Google Scholar] [CrossRef][Green Version]
- Reed, D.H.; Frankham, R. Correlation between Fitness and Genetic Diversity. Conserv. Biol. 2003, 17, 230–237. [Google Scholar] [CrossRef]
- Hoffman, J.R.; Willoughby, J.R.; Swanson, B.J.; Pangle, K.L.; Zanatta, D.T. Detection of Barriers to Dispersal Is Masked by Long Lifespans and Large Population Sizes. Ecol. Evol. 2017, 7, 9613–9623. [Google Scholar] [CrossRef]
- Froufe, E.; Duarte, D.V.; Teixeira, A.; Sousa, R.; Varandas, S.; Ghamizi, M.; Zieritz, A.; Lopes-Lima, M. Who Lives Where? Molecular and Morphometric Analyses Clarify Which Unio Species (Unionida, Mollusca) Inhabit the Southwestern Palearctic. Org. Divers. Evol. 2016, 43500, 597–611. [Google Scholar] [CrossRef]
- Zieritz, A.; Froufe, E.; Bolotov, I.; Gonçalves, D.V.; Aldridge, D.C.; Bogan, A.E.; Gan, H.M.; Gomes-Dos-Santos, A.; Sousa, R.; Teixeira, A.; et al. Mitogenomic Phylogeny and Fossil-Calibrated Mutation Rates for All F- and M-Type MtDNA Genes of the Largest Freshwater Mussel Family, the Unionidae (Bivalvia). Zool. J. Linn. Soc. 2021, 193, 1088–1107. [Google Scholar] [CrossRef]
- Mangerud, J.; Jakobsson, M.; Alexanderson, H.; Astakhov, V.; Clarke, G.K.C.; Henriksen, M.; Hjort, C.; Krinner, G.; Lunkka, J.P.; Möller, P.; et al. Ice-Dammed Lakes and Rerouting of the Drainage of Northern Eurasia during the Last Glaciation. Quat. Sci. Rev. 2004, 23, 1313–1332. [Google Scholar] [CrossRef]
- Krinner, G.; Mangerud, J.; Jakobsson, M.; Crucifix, M.; Ritz, C.; Svendsen, J.I. Enhanced Ice Sheet Growth in Eurasia Owing to Adjacent Ice-Dammed Lakes. Nature 2004, 427, 429–432. [Google Scholar] [CrossRef] [PubMed]
- Bernatchez, L. The Evolutionary History of Brown Trout (Salmo trutta L.) Inferred from Phylogeographic, Nested Clade, and Mismatch Analyses of Mitochondrial DNA Variation. Evolution 2001, 55, 351–379. [Google Scholar] [CrossRef] [PubMed]
- Treasurer, J.W.; Turnbull, T. The Pathology and Seawater Performance of Farmed Atlantic Salmon Infected with Glochidia of Margaritifera margaritifera. J. Fish Biol. 2000, 57, 858–866. [Google Scholar] [CrossRef]
- Satkūnas, J.; Grigienė, A.; Jusienė, A.; Damušytė, A.; Mažeika, J. Pleni-weichselian Sequences in the Venta River Valley and Vicinities (North-Western Lithuania), Exemplified by the Purviai Outcrop. In Proceedings of the Quaternary of Western Lithuania: From the Pleistocene Glaciations to the Evolution of the Baltic Sea: The INQUA Peribaltic Group Field Symposium, Plateliai, Lithuania, 27 May–2 June 2007; Guobytė, R., Stančikaitė, M., Eds.; Lithuanian Geological Survey, Institute of Geology and Geography: Vilnius, Lithuania, 2007; pp. 71–72. [Google Scholar]
- Artamonova, V.S.; Bolotov, I.N.; Vinarski, M.V.; Makhrov, A.A. Fresh-and Brackish-Water Cold-Tolerant Species of Southern Europe: Migrants from the Paratethys That Colonized the Arctic. Water 2021, 13, 1161. [Google Scholar] [CrossRef]
- Kazakov, R.V.; Titov, S.F. Geographical Patterns in the Population Genetics of Atlantic Salmon, Salmo salar L., on U.S.S.R. Territory, as Evidence for Colonization Routes. J. Fish Biol. 1991, 39, 1–6. [Google Scholar] [CrossRef]
- Cortey, M.; Vera, M.; Pla, C.; GarcÍa-MarÍn, J.L. Northern and Southern Expansions of Atlantic Brown Trout (Salmo trutta) Populations during the Pleistocene. Biol. J. Linn. Soc. 2009, 97, 904–917. [Google Scholar] [CrossRef]
- Zueva, K.J. Evolutionary Genomics of Adaptation in Atlantic Salmon From Northern Europe. Ph.D. Thesis, University of Turku, Turku, Finland, 2021. [Google Scholar]
- Johansson, P. Late Weichselian Deglaciation in Finnish Lapland. Appl. Quat. Res. Cent. Part Glaciat. Terrain 2007, 2007, 47–54. [Google Scholar]
- Johansson, P.; Lunkka, J.P.; Sarala, P. Late Pleistocene Glacigenic Deposits from the Central Part of the Scandinavian Ice Sheet to Younger Dryas End Moraine Zone. In Proceedings of the INQUA Peribaltic Working Group Meeting and Excursion in Finland, Kilpisjärvi, Finland, 12–17 June 2011; Johansson, P., Lunkka, J.-P., Sarala, P., Eds.; Geological Survey of Finland: Rovaniemi, Finland, 2011. ISBN 9789522171634. [Google Scholar]
- Elina, G.A.; Lukashov, A.D.; Yurkovskaya, T.K. Late Glacial and Holocene Time in the East Fennoscandia (Palaeovegetation and Palaeogeography); Karelian Research Centre: Petrozavodsk, Russia, 2000. [Google Scholar]
- Ziuganov, V.; Kaliuzhin, S.; Beletsky, V.; Popkovitch, E. The Pearl Mussel-Salmon Community in the Varzuga River, Northwest Russia: Problems of Environmental Impacts. In Ecology and Evolution of the freshwater mussels Unionoida; Springer: Berlin/Heidelberg, Germany, 2001; Volume 145, pp. 359–366. [Google Scholar] [CrossRef]
- Ieshko, E.P.; Geist, J.; Murzina, S.A.; Veselov, A.E.; Lebedeva, D.I.; Ziuganov, V.V. The Characteristics of the Infection of Juvenile Atlantic Salmon with Glochidia of the Freshwater Pearl Mussel in Rivers of Northwest Russia. Knowl. Manag. Aquat. Ecosyst. 2016, 6, 10. [Google Scholar] [CrossRef]
- Popov, I. In the Search of the Lost Pearl; Springer International Publishing: Cham, Switzerland, 2021. [Google Scholar]
- Popov, I.Y. Overfishing in the Baltic Sea basin in Russia, its impact on the pearl mussel, and possibilities for the conservation of riverine ecosystems in conditions of high anthropogenic pressure. Biol. Bull. 2017, 44, 39–44. [Google Scholar] [CrossRef]
- Araujo, R.; Schneider, S.; Roe, K.J.; Erpenbeck, D.; Machordom, A. The origin and phylogeny of Margaritiferidae (Bivalvia, Unionoida): A synthesis of molecular and fossil data. Zool. Scr. 2016, 46, 289–307. [Google Scholar] [CrossRef]
- Gomes-Dos-Santos, A.; Lopes-Lima, M.; Castro, L.F.C.; Froufe, E. Molluscan genomics: The road so far and the way forward. Hydrobiologia 2019, 847, 1705–1726. [Google Scholar] [CrossRef]
- Huff, S.W.; Campbell, D.; Gustafson, D.L.; Lydeard, C.; Altaba, C.R.; Giribet, G. Investigations into the Phylogenetic Relationships of Freshwater Pearl Mussels (Bivalvia: Margaritiferidae) Based on Molecular Data: Implications for Their Taxonomy and Biogeography. J. Molluscan Stud. 2004, 70, 379–388. [Google Scholar] [CrossRef]
- Källersjö, M.; Von Proschwitz, T.; Lundberg, S.; Eldenäs, P.; Erséus, C. Evaluation of ITS RDNA as a Complement to Mitochondrial Gene Sequences for Phylogenetic Studies in Freshwater Mussels: An Example Using Unionidae from North-Western Europe. Zool. Scr. 2005, 34, 415–424. [Google Scholar] [CrossRef]
- LeBlanc, F.; Steeves, R.; Belliveau, V.; Akaishi, F.; Gagné, N. Detecting the brook floater, a freshwater mussel species at risk, using environmental DNA. Aquat. Conserv. Mar. Freshw. Ecosyst. 2021, 31, 1233–1244. [Google Scholar] [CrossRef]
- Sharma, P.P.; Zardus, J.D.; Boyle, E.E.; González, V.L.; Jennings, R.; McIntyre, E.; Wheeler, W.C.; Etter, R.J.; Giribet, G. Into the deep: A phylogenetic approach to the bivalve subclass Protobranchia. Mol. Phylogenet. Evol. 2013, 69, 188–204. [Google Scholar] [CrossRef]
- Vikhrev, I.V.; Bolotov, I.N.; Altun, A.; Gofarov, M.; Dvoryankin, G.A.; Kondakov, A.; Ozcan, T.; Ozcan, G. The revenant: Rediscovery of Margaritifera homsensis from Orontes drainage with remarks on its taxonomic status and conservation (Bivalvia: Margaritiferidae). Syst. Biodivers. 2017, 16, 69–80. [Google Scholar] [CrossRef]
- Whelan, N.V.; Geneva, A.J.; Graf, D.L. Molecular phylogenetic analysis of tropical freshwater mussels (Mollusca: Bivalvia: Unionoida) resolves the position of Coelatura and supports a monophyletic Unionidae. Mol. Phylogenet. Evol. 2011, 61, 504–514. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No | Population | n | NH | Hd ± SD | π ± SD | Ramos and Onsis’s R2-Test | Fu’s FS-Test | Tajima’s D-Test | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
| R2 | p-Value | FS | p-Value | D | p-Value | ||||||
| 1 | Varzuga | 8 | 4 | 0.82 ±0.10 | 0.004 ± 0.002 | 0.21 | 0.78 | 0.79 | 0.680 | 0.81 | 0.810 |
| 2 | Mutkajoki | 23 | 3 | 0.54 ±0.10 | 0.002 ± 0.002 | 0.17 | 0.77 | 2.21 | 0.870 | 0.75 | 0.770 |
| 3 | Nuris | 7 | 2 | 0.29 ±0.20 | 0.001 ± 0.001 | 0.35 | 0.70 | −0.09 | 0.240 | −1.00 | 0.230 |
| 4 | Tavajoki | 20 | 2 | 0.52 ±0.04 | 0.001 ± 0.001 | 0.26 | 1.00 | 1.46 | 0.690 | 1.53 | 0.960 |
| 5 | Tuhka | 8 | 2 | 0.25 ±0.18 | 0.0004 ± 0.001 | 0.33 | 0.74 | −0.18 | 0.200 | −1.05 | 0.220 |
| 6 | Vozhma | 15 | 7 | 0.86 ± 0.06 | 0.003 ± 0.002 | 0.12 | 0.30 | −2.00 | 0.080 | −0.38 | 0.400 |
| 7 | Ukhta | 14 | 5 | 0.7 ± 0.10 | 0.002 ± 0.001 | 0.16 | 0.62 | −0.83 | 0.330 | −1.02 | 0.160 |
| 8 | Lopshenga | 28 | 4 | 0.65 ± 0.06 | 0.003 ± 0.002 | 0.2 | 0.95 | 1.65 | 0.800 | 1.51 | 0.920 |
| 9 | Kem | 30 | 10 | 0.88 ± 0.03 | 0.003 ± 0.002 | 0.11 | 0.43 | −2.80 | 0.080 | −0.45 | 0.360 |
| 10 | Vuokkijoki | 22 | 10 | 0.83 ± 0.06 | 0.004 ± 0.002 | 0.13 | 0.47 | −3.38 | 0.027 | −0.73 | 0.250 |
| 11 | Solza | 30 | 8 | 0.82 ± 0.05 | 0.004 ± 0.002 | 0.13 | 0.70 | −0.14 | 0.527 | 0.18 | 0.620 |
| 12 | Maloshuika | 30 | 5 | 0.7 ± 0.07 | 0.003 ± 0.002 | 0.19 | 0.89 | 0.47 | 0.620 | 1.26 | 0.900 |
| 13 | Nimenga | 30 | 8 | 0.8 ± 0.04 | 0.004 ± 0.002 | 0.11 | 0.47 | −0.83 | 0.350 | −0.30 | 0.400 |
| 14 | Kozha | 15 | 6 | 0.86 ± 0.05 | 0.002 ± 0.002 | 0.14 | 0.47 | −1.53 | 0.120 | 0.01 | 0.600 |
| 15 | Yanega | 30 | 5 | 0.54 ± 0.10 | 0.002 ± 0.001 | 0.1 | 0.36 | −0.05 | 0.470 | −0.51 | 0.380 |
| 16 | Peypia | 30 | 1 | 0 | 0 | n/a | n/a | n/a | |||
| 17 | Khorinka | 30 | 3 | 0.35 ± 0.103 | 0.001 ± 0.001 | 0.09 | 0.18 | −0.50 | 0.200 | −0.50 | 0.330 |
| Population group | |||||||||||
| BS | 99 | 10 | 0.62 ± 0.05 | 0.002 ± 0.001 | 0.05 | 0.07 | −5.26 | 0.027 | −1.41 | 0.060 | |
| WNA | 10 | 6 | 0.89 ± 0.07 | 0.004 ± 0.002 | 0.13 | 0.18 | −1.51 | 0.117 | −1.26 | 0.110 | |
| WS | 317 | 27 | 0.88 ± 0.01 | 0.003 ± 0.002 | 0.05 | 0.12 | −12.66 | 0.002 | −1.01 | 0.160 | |
| BRIT | 11 | 3 | 0.56 ± 0.13 | 0.004 ± 0.003 | 0.21 | 0.83 | 2.87 | 0.930 | 1.03 | 0.860 | |
| IBER | 20 | 3 | 0.35 ± 0.12 | 0.001 ± 0.001 | 0.13 | 0.47 | −0.77 | 0.200 | −0.81 | 0.235 | |
| Structure Tested | Source of Variation | % Variance | Fixation Index | p |
|---|---|---|---|---|
| Basin | Among groups | 24.23 | 0.190 | <0.001 |
| Among populations within groups | 14.32 | 0.390 | <0.001 | |
| Within populations | 61.46 | 0.240 | <0.001 | |
| Host | Among groups | −0.45 | 0.210 | <0.001 |
| Among populations within groups | 20.64 | 0.200 | <0.001 | |
| Within populations | 79.81 | −0.006 | 0.48 |
| Group of Populations | BRIT | BS | IBER | WNA | WS |
|---|---|---|---|---|---|
| BRIT | 0 | 0.51829 | 0.30692 | 0.25387 | 0.30222 |
| BS | 0 | 0.76573 | 0.02165 | 0.06705 | |
| IBER | 0 | 0.72199 | 0.55286 | ||
| WNA | 0 | 0.00520 | |||
| WS | 0 |
| Response Variable | Source | d.f | SS | F | p |
|---|---|---|---|---|---|
| PC1 | Host | - | - | - | n.s. |
| Basin | 1 | 317.762 | 5.282 | 0.04 | |
| Error | 14 | 842.306 | |||
| PC2 | Host | - | - | - | n.s. |
| Basin | 1 | 116.615 | 4.918 | 0.04 | |
| Error | 14 | 331.983 | |||
| PC6 | Host | 1 | 21.157 | 6.439 | 0.02 |
| Basin | - | - | - | n.s. | |
| Error | 14 | 46.001 |
| PC | Eigenvalue | % Variance |
|---|---|---|
| 1 | 84.6624 | 51.3110 |
| 2 | 28.8112 | 17.4610 |
| 3 | 18.8291 | 11.4120 |
| 4 | 15.3443 | 9.2995 |
| 5 | 7.2902 | 4.4183 |
| 6 | 4.2221 | 2.5588 |
| 7 | 2.0581 | 1.2473 |
| 8 | 1.3401 | 0.8122 |
| 9 | 0.9264 | 0.5615 |
| 10 | 0.6488 | 0.3932 |
| 11 | 0.3759 | 0.2278 |
| 12 | 0.2849 | 0.1726 |
| 13 | 0.1383 | 0.0838 |
| 14 | 0.0546 | 0.0331 |
| 15 | 0.0138 | 0.0084 |
| 16 | 0.0001 | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vikhrev, I.V.; Ieshko, E.P.; Kondakov, A.V.; Mugue, N.S.; Bovykina, G.V.; Efremov, D.A.; Bulakhov, A.G.; Tomilova, A.A.; Yunitsyna, O.A.; Bolotov, I.N. Postglacial Expansion Routes and Mitochondrial Genetic Diversification of the Freshwater Pearl Mussel in Europe and North America. Diversity 2022, 14, 477. https://doi.org/10.3390/d14060477
Vikhrev IV, Ieshko EP, Kondakov AV, Mugue NS, Bovykina GV, Efremov DA, Bulakhov AG, Tomilova AA, Yunitsyna OA, Bolotov IN. Postglacial Expansion Routes and Mitochondrial Genetic Diversification of the Freshwater Pearl Mussel in Europe and North America. Diversity. 2022; 14(6):477. https://doi.org/10.3390/d14060477
Chicago/Turabian StyleVikhrev, Ilya V., Evgenii P. Ieshko, Alexander V. Kondakov, Nikolai S. Mugue, Galina V. Bovykina, Denis A. Efremov, Andrei G. Bulakhov, Alena A. Tomilova, Olesya A. Yunitsyna, and Ivan N. Bolotov. 2022. "Postglacial Expansion Routes and Mitochondrial Genetic Diversification of the Freshwater Pearl Mussel in Europe and North America" Diversity 14, no. 6: 477. https://doi.org/10.3390/d14060477
APA StyleVikhrev, I. V., Ieshko, E. P., Kondakov, A. V., Mugue, N. S., Bovykina, G. V., Efremov, D. A., Bulakhov, A. G., Tomilova, A. A., Yunitsyna, O. A., & Bolotov, I. N. (2022). Postglacial Expansion Routes and Mitochondrial Genetic Diversification of the Freshwater Pearl Mussel in Europe and North America. Diversity, 14(6), 477. https://doi.org/10.3390/d14060477