
Genetic Structure and Demographic History of Yellow Grouper (Epinephelus awoara) from the Coast of Southeastern Mainland China, Inferred by Mitochondrial, Nuclear and Microsatellite DNA Markers


Abstract
1. Introduction
2. Materials and Methods
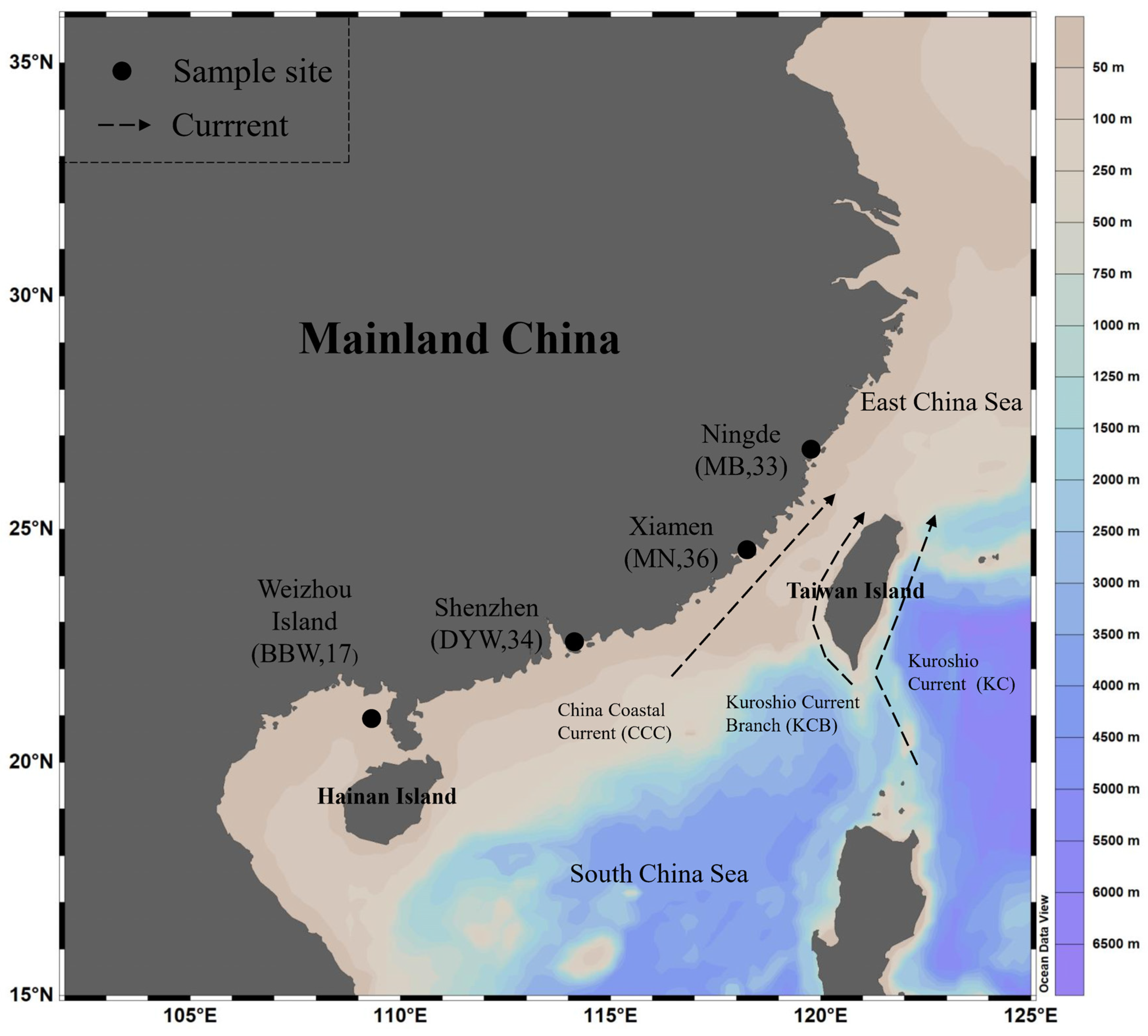
2.1. Sample Collection, Microsatellite Genotyping, and Mitochondrial and Nuclear Sequencing
2.2. Data Analysis
2.2.1. Sequence Analysis
2.2.2. Microsatellite DNA Analysis
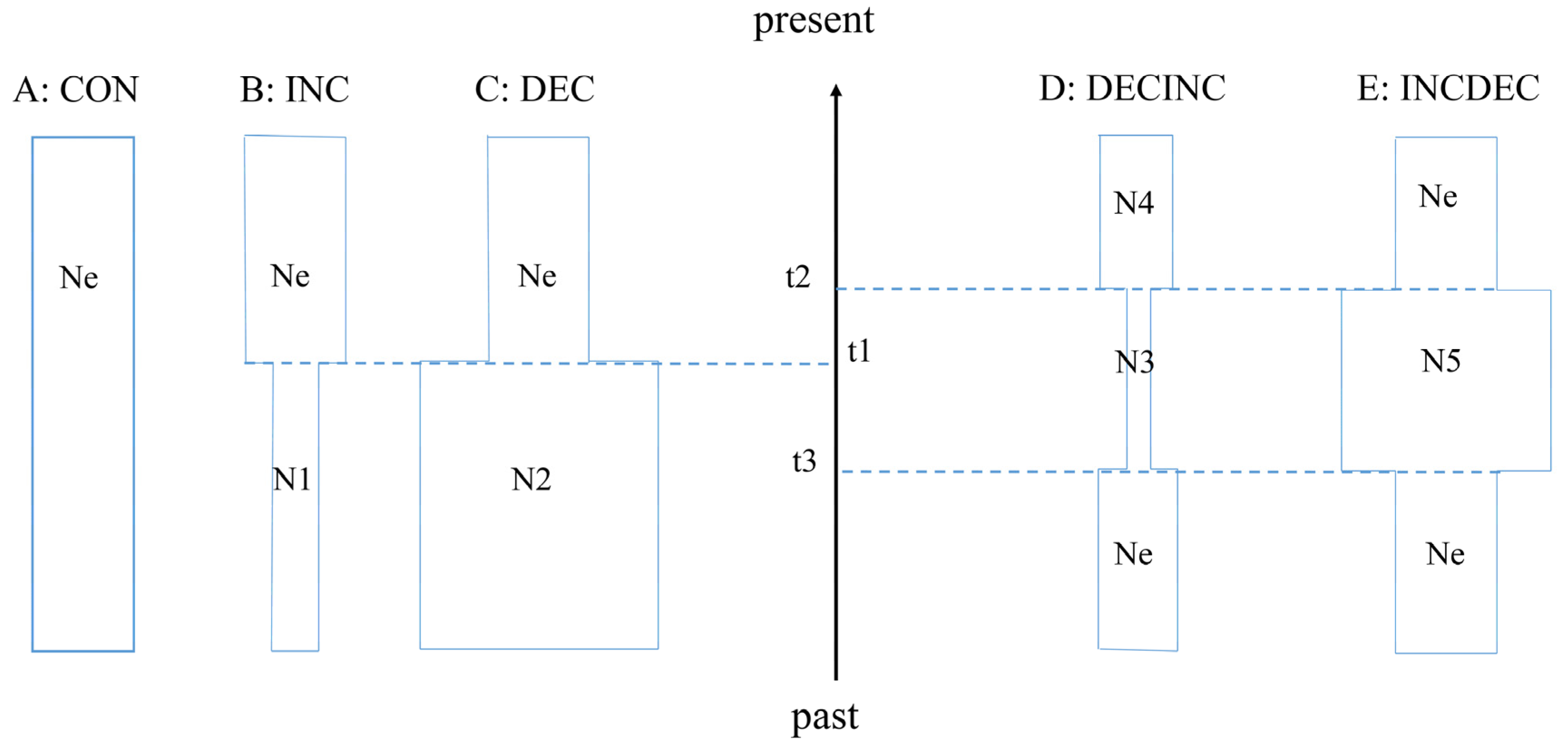
2.2.3. DIY-ABC Analysis
3. Result
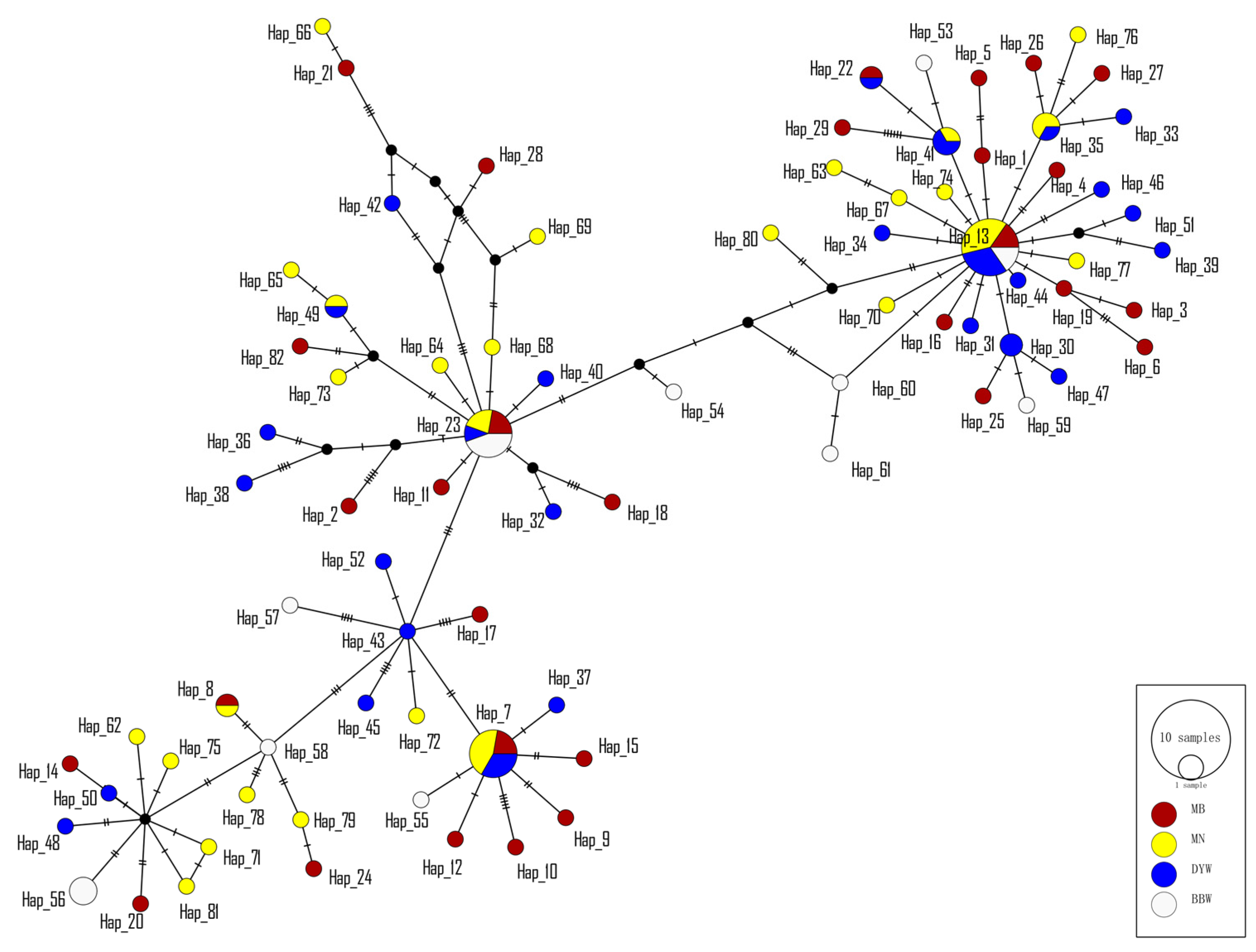
3.1. Mitochondrial DNA
3.2. Nuclear DNA
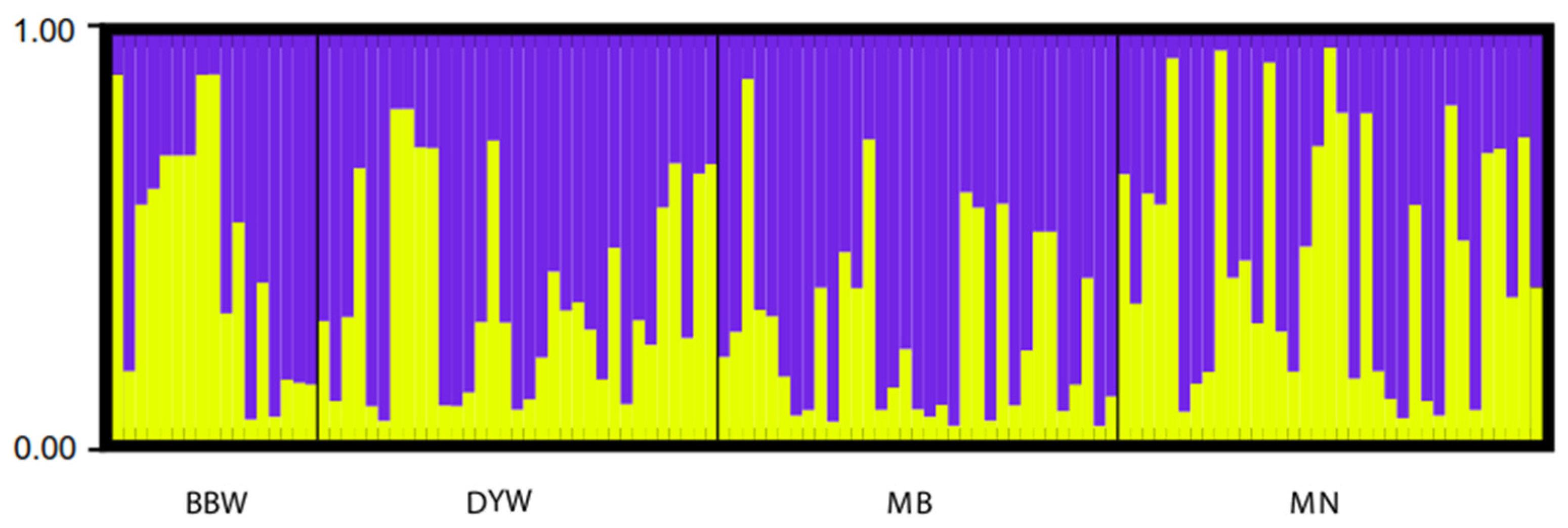
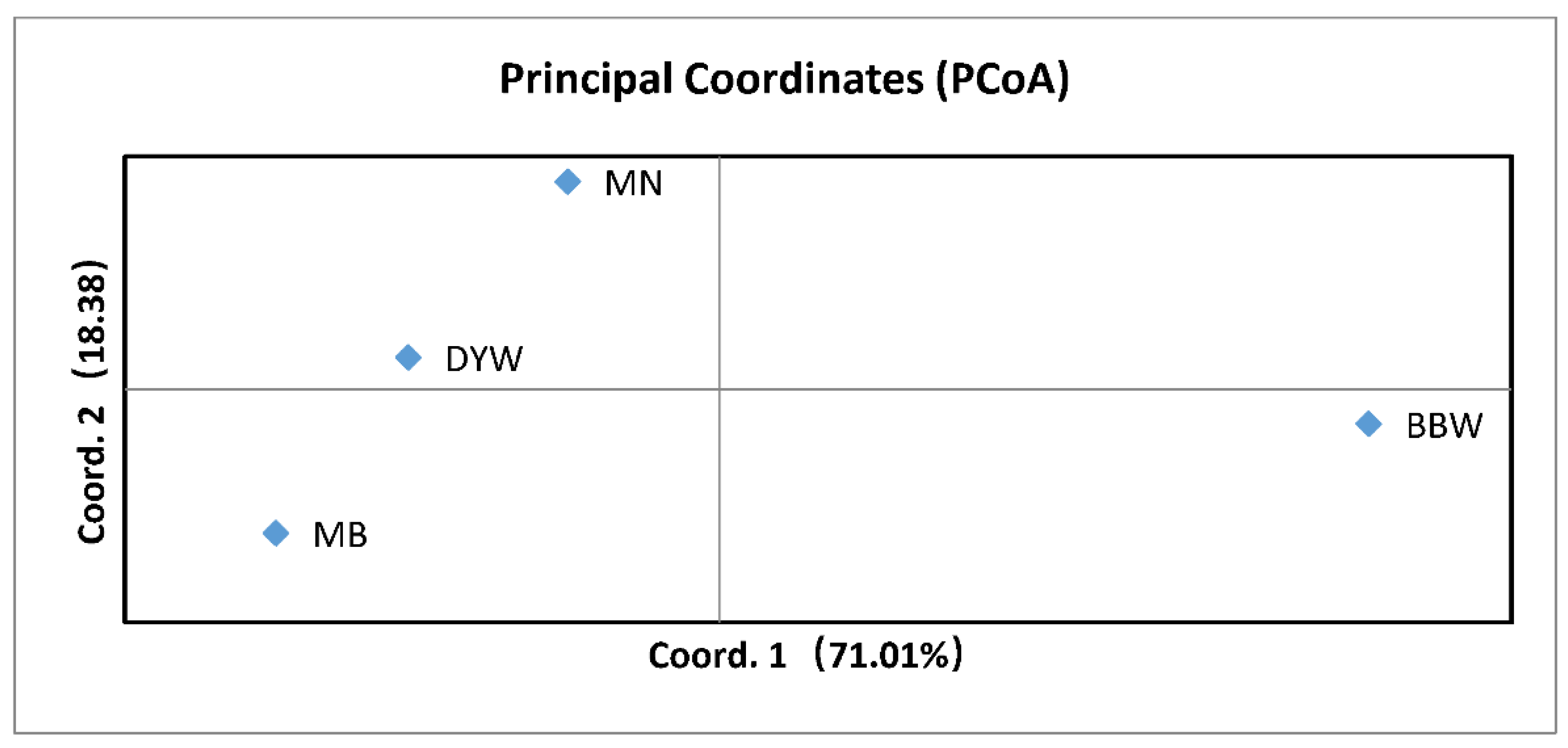
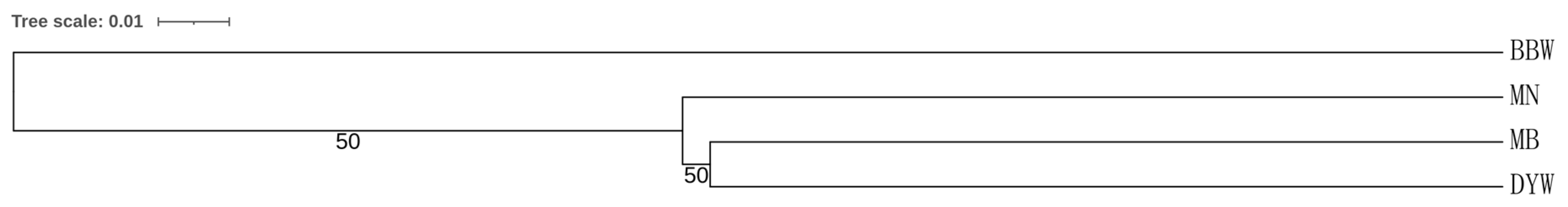
3.3. Microsatellite DNA
4. Discussion
4.1. Genetic Diversity
4.2. Population Structure
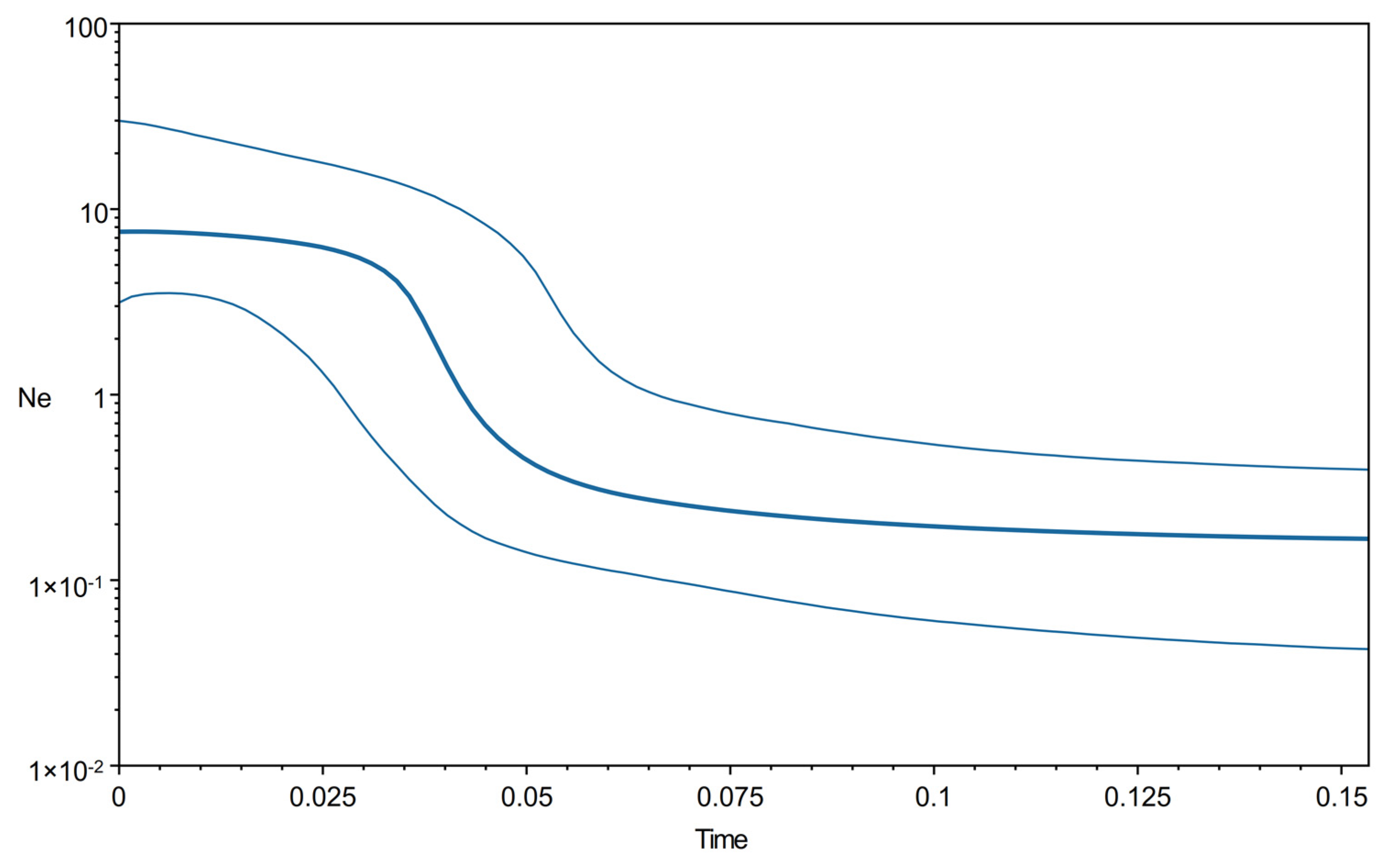
4.3. Demographic History and DIY-ABC
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Avise, J.C.; Arnold, J.; Ball, R.M.; Bermingham, E.; Lamb, T.; Neigel, J.E.; Reeb, C.A.; Saunders, N.C. Intraspecific Phylogeography: The Mitochondrial DNA Bridge Between Population Genetics and Systematics. Annu. Rev. Ecol. Evol. Syst. 1987, 18, 489–522. [Google Scholar] [CrossRef]
- Hewitt, G.M. Genetic consequences of climatic oscillations in the Quaternary. Philos. Trans. R. Soc. B—Biol. Sci. 2004, 359, 183–195. [Google Scholar] [CrossRef] [PubMed]
- Teske, P.R.; von der Heyden, S.; McQuaid, C.D.; Barker, N.P. A review of marine phylogeography in southern Africa. S. Afr. J. Sci. 2011, 107, 43–53. [Google Scholar] [CrossRef]
- Ibrahim, K.M.; Nichols, R.A.; Hewitt, G.M. Spatial patterns of genetic variation generated by different forms of dispersal during range expansion. Heredity 1996, 77, 282–291. [Google Scholar] [CrossRef]
- Bohonak, A.J. Dispersal, gene flow, and population structure. Q. Rev. Biol. 1999, 74, 21–45. [Google Scholar] [CrossRef]
- Liu, B.J.; Zhang, B.D.; Xue, D.X.; Gao, T.X.; Liu, J.X. Population Structure and Adaptive Divergence in a High Gene Flow Marine Fish: The Small Yellow Croaker (Larimichthys polyactis). PLoS ONE 2016, 11, e0144637. [Google Scholar] [CrossRef]
- Barber, P.H.; Palumbi, S.R.; Erdmann, M.V.; Moosa, M.K. Sharp genetic breaks among populations of Haptosquilla pulchella (Stomatopoda) indicate limits to larval transport: Patterns, causes, and consequences. Mol. Ecol. 2002, 11, 659–674. [Google Scholar] [CrossRef]
- Benzie, J.A.H. Major genetic differences between crown-of-thorns starfish (Acanthaster planci) populations in the Indian and Pacific Oceans. Evolution 1999, 53, 1782–1795. [Google Scholar] [CrossRef]
- Selkoe, K.A.; Toonen, R.J. Marine connectivity: A new look at pelagic larval duration and genetic metrics of dispersal. Mar. Ecol. Prog. Ser. 2011, 436, 291–305. [Google Scholar] [CrossRef]
- White, C.; Selkoe, K.A.; Watson, J.; Siegel, D.A.; Zacherl, D.C.; Toonen, R.J. Ocean currents help explain population genetic structure. Proc. R. Soc. B—Biol. Sci. 2010, 277, 1685–1694. [Google Scholar] [CrossRef]
- Kingsford, M.J.; Leis, J.M.; Shanks, A.; Lindeman, K.C.; Morgan, S.G.; Pineda, J. Sensory environments, larval abilities and local self-recruitment. Bull. Mar. Sci. 2002, 70, 309–340. [Google Scholar]
- Planes, S.; Fauvelot, C. Isolation by distance and vicariance drive genetic structure of a coral reef fish in the Pacific Ocean. Evolution 2002, 56, 378–399. [Google Scholar] [CrossRef] [PubMed]
- Jamaludin, N.A.; Mohd-Arshaad, W.; Akib, N.A.M.; Abidin, D.H.Z.; Nghia, N.V.; Nor, S.M. Phylogeography of the Japanese scad, Decapterus maruadsi (Teleostei; Carangidae) across the Central Indo-West Pacific: Evidence of strong regional structure and cryptic diversity. Mitochondrial DNA Part A 2020, 31, 298–310. [Google Scholar] [CrossRef] [PubMed]
- Salas, E.; Bernardi, G.; Berumen, M.L.; Gaither, M.; Rocha, L. RADseq analyses reveal concordant Indian Ocean biogeographic and phylogeographic boundaries in the reef fish Dascyllus trimaculatus. R. Soc. Open Sci. 2019, 6, 172413. [Google Scholar] [CrossRef]
- Tamaki, K.; Honza, E. Global tectonics and formation of marginal basins—Role of the western pacific. Episodes 1991, 14, 224–230. [Google Scholar] [CrossRef]
- Liu, J.X.; Gao, T.X.; Yokogawa, K.; Zhang, Y.P. Differential population structuring and demographic history of two closely related fish species, Japanese sea bass (Lateolabrax japonicus) and spotted sea bass (Lateolabrax maculatus) in Northwestern Pacific. Mol. Phylogenet. Evol. 2006, 39, 799–811. [Google Scholar] [CrossRef]
- Wang, P. Response of Western Pacific marginal seas to glacial cycles: Paleoceanographic and sedimentological features. Mar. Geol. 1999, 156, 5–39. [Google Scholar] [CrossRef]
- Chen, S.P.; Liu, T.; Li, Z.F.; Gao, T.X. Genetic population structuring and demographic history of red spotted grouper (Epinephelus akaara) in South and East China Sea. Afr. J. Biotechnol. 2008, 7, 3554–3562. [Google Scholar]
- Antoro, S.; Na-Nakorn, U.; Koedprang, W. Study of genetic diversity of orange-spotted grouper, Epinephelus coioides, from Thailand and Indonesia using microsatellite markers. Mar. Biotechnol. 2006, 8, 17–26. [Google Scholar] [CrossRef]
- Li, Y.; Han, Z.; Song, N.; Gao, T.X. New evidence to genetic analysis of small yellow croaker (Larimichthys polyactis) with continuous distribution in China. Biochem. Syst. Ecol. 2013, 50, 331–338. [Google Scholar] [CrossRef]
- Yan, S.; Catanese, G.; Brown, C.L.; Wang, M.; Yang, C.; Yang, T. Phylogeographic study on the chub mackerel (Scomber japonicus) in the Northwestern Pacific indicates the late Pleistocene population isolation. Mar. Ecol. 2015, 36, 753–765. [Google Scholar] [CrossRef]
- Palumbi, S. Molecular biogeography of the Pacific. Coral Reefs 1997, 16, S47–S52. [Google Scholar] [CrossRef]
- Qiu, F.; Li, H.; Lin, H.; Ding, S.; Miyamoto, M.M. Phylogeography of the inshore fish, Bostrychus sinensis, along the Pacific coastline of China. Mol. Phylogenet. Evol. 2016, 96, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Ding, S.; Mishra, M.; Wu, H.; Liang, S.; Miyamoto, M.M. Characterization of hybridization within a secondary contact region of the inshore fish, Bostrychus sinensis, in the East China Sea. Heredity 2018, 120, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Xu, H.; Chen, Z.; Wang, Y. Marine fish cage culture in China. The Future of Mariculture: A Regional Approach for Responsible Development in the Asia-Pacific Region. 2008; p. 285. Available online: https://www.fao.org/3/i0202e/i0202e14.pdf (accessed on 19 August 2021).
- Coker, D.J.; DiBattista, J.D.; Sinclair-Taylor, T.H.; Berumen, M.L. Spatial patterns of cryptobenthic coral-reef fishes in the Red Sea. Coral Reefs 2018, 37, 193–199. [Google Scholar] [CrossRef]
- Dooley, J. A Checklist of Fishes of the South China Sea: Malacanthidae. In A Checklist of Fishes of the South China Sea; National University of Singapore: Singapore, 2000; pp. 569–667. [Google Scholar]
- Lindberg, G.U.; Krasyukova, Z.V. Fishes of the Sea of Japan and the Adjacent Areas of the Sea of Okhotsk and the Yellow Sea; CRC Press: Boca Raton, FL, USA, 2020. [Google Scholar]
- To, A.W.; Shea, S.K. Rapid Reef Fish Survey at Daya Bay, Guangdong Province, China; City University of Hong Kong: Hong Kong, 2015. [Google Scholar]
- Hodgkiss, I. Fifty common marine food fishes of Hong Kong. Mem. Hong Kong Nat. Hist. Soc. 1988, 18, 19–33. [Google Scholar]
- Cornish, A.S. The reef fish species of the Cape Daguilar Marine Reserve, Hoi Ha Wan Marine Park, Yan Chau Tong Marine Park and Ping Chau, Hong Kong. Mar. Flora Fauna Hong Kong S. China V 2000, 5, 369. [Google Scholar]
- Heemstra, P.C.; Randall, J.E. Groupers of the world. FAO Fish. Synop. 1993, 16, 1. [Google Scholar]
- Sun, L.; Chen, H.; Huang, L. Growth, faecal production, nitrogenous excretion and energy budget of juvenile yellow grouper (Epinephelus awoara) relative to ration level. Aquaculture 2007, 264, 228–235. [Google Scholar] [CrossRef]
- Zhou, Z.G.; Liu, Y.C.; Shi, P.J.; He, S.X.; Yao, B.; Ringo, E. Molecular characterization of the autochthonous microbiota in the gastrointestinal tract of adult yellow grouper (Epinephelus awoara) cultured in cages. Aquaculture 2009, 286, 184–189. [Google Scholar] [CrossRef]
- Xu, X.J.; Sang, B.H.; Luo, G. A genetic role for macrophage migration inhibitory factor (MIF) in Epinephelus awoara infected with Vibrio parahaemolyticus. Eur. J. Inflamm. 2016, 14, 184–189. [Google Scholar] [CrossRef]
- Doyle, M.; Morse, W.; Jr, A. A Comparison of Larval Fish Assemblages in the Temperate Zone of the Northeast Pacific and Northwest Atlantic Oceans. Bull. Mar. Sci. 1993, 53, 588–644. [Google Scholar]
- Kim, W.J.; Kim, K.K.; Han, H.S.; Nam, B.H.; Kim, Y.O.; Kong, H.J.; Noh, J.K.; Yoon, M. Population structure of the olive flounder (Paralichthys olivaceus) in Korea inferred from microsatellite marker analysis. J. Fish Biol. 2010, 76, 1958–1971. [Google Scholar] [CrossRef] [PubMed]
- Umino, T.; Kajihara, T.; Shiozaki, H.; Ohkawa, T.; Jeong, D.-S.; Ohara, K. Wild stock structure of Girella punctata in Japan revealed shallow genetic differentiation but subtle substructure in subsidiary distributions. Fish. Sci. 2009, 75, 909–919. [Google Scholar] [CrossRef]
- Buchholz-Sørensen, M.; Vella, A. Population structure, genetic diversity, effective population size, demographic history and regional connectivity patterns of the endangered dusky grouper, Epinephelus marginatus (Teleostei: Serranidae), within Malta’s fisheries management zone. PLoS ONE 2016, 11, e0159864. [Google Scholar] [CrossRef] [PubMed]
- Upadhyay, S.K.; Jun, W.; Yong-Ouan, S.; Shao-Xiong, D.; Chaturvedi, S. Genetic diversity of yellow grouper (Epinephelus awoara) determined by random amplified polymorphic DNA (RAPD) analysis. Fish. Bull. 2006, 104, 638–642. [Google Scholar]
- Zhao, L.; Shao, C.; Liao, X.; Ma, H.; Zhu, X.; Chen, S. Twelve novel polymorphic microsatellite loci for the Yellow grouper (Epinephelus awoara) and cross-species amplifications. Conserv. Genet. 2009, 10, 743–745. [Google Scholar] [CrossRef]
- Karim, A.A.; Norziah, M.H.; Seow, C.C. Methods for the study of starch retrogradation. Food Chem. 2000, 71, 9–36. [Google Scholar] [CrossRef]
- Arias, M.C.; Aulagnier, S.; Baerwald, E.F.; Barclay, R.M.; Batista, J.S.; Beasley, R.R.; Bezerra, R.A.; Blanc, F.; Bridge, E.S.; Cabria, M.T. Microsatellite records for volume 8, issue 1. Conserv. Genet. Resour. 2016, 8, 43–81. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Dufayard, J.-F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Huelsenbeck, J.P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 2003, 19, 1572–1574. [Google Scholar] [CrossRef] [PubMed]
- Lefort, V.; Longueville, J.-E.; Gascuel, O. SMS: Smart model selection in PhyML. Mol. Biol. Evol. 2017, 34, 2422–2424. [Google Scholar] [CrossRef] [PubMed]
- Leigh, J.; Bryant, D. PopART: Full-Feature Software for Haplotype Network Construction. Methods Ecol. Evol. 2015, 6, 1110–1116. [Google Scholar] [CrossRef]
- Excoffier, L.; Lischer, H.E.L. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar] [CrossRef] [PubMed]
- Bohonak, A.J. IBD (isolation by distance): A program for analyses of isolation by distance. J. Hered. 2002, 93, 153–154. [Google Scholar] [CrossRef] [PubMed]
- Tajima, F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 1989, 123, 585–595. [Google Scholar] [CrossRef]
- Fu, Y.-X. Statistical Tests of Neutrality of Mutations Against Population Growth, Hitchhiking and Background Selection. Genetics 1997, 147, 915–925. [Google Scholar] [CrossRef]
- Drummond, A.J.; Suchard, M.A.; Xie, D.; Rambaut, A. Bayesian Phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 2012, 29, 1969–1973. [Google Scholar] [CrossRef]
- Bouckaert, R.; Heled, J.; Kühnert, D.; Vaughan, T.; Wu, C.-H.; Xie, D.; Suchard, M.A.; Rambaut, A.; Drummond, A.J. BEAST 2: A software platform for Bayesian evolutionary analysis. PLoS Comput. Biol. 2014, 10, e1003537. [Google Scholar] [CrossRef] [PubMed]
- Kumazawa, Y. Molecular clock estimation in fishes and its application to biogeographical studies. Fish. Sci. 2002, 68, 357–360. [Google Scholar] [CrossRef][Green Version]
- Bermingham, E.; McCafferty, S.S.; Martin, A. Fish biogeography and molecular clocks: Perspectives from the Panamanian Isthmus. Mol. Syst. Fishes 1997, 113–128. [Google Scholar]
- Van Oosterhout, C.; Hutchinson, W.F.; Wills, D.P.M.; Shipley, P. Micro-Checker: Software for identifying and correcting genotyping errors in microsatellite data. Mol. Ecol. Notes 2004, 4, 535–538. [Google Scholar] [CrossRef]
- Goudet, J. FSTAT (Version 2.9.3): A Program to Estimate and Test Gene Diversities and Fixation Indices. 2001. Available online: www.unil.ch/izea/softwares/fstat.html (accessed on 19 August 2021).
- Weir, B.S.; Cockerham, C.C. estimating f-statistics for the analysis of population-structure. Evolution 1984, 38, 1358–1370. [Google Scholar] [CrossRef]
- Earl, D.A.; Vonholdt, B.M. Structure Harvester: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Peakall, R.; Smouse, P.E. GenAlEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research-an update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef]
- Nei, M. Genetic Distance between Populations. Am. Nat. 1972, 106, 283–292. [Google Scholar] [CrossRef]
- Piry, S.; Luikart, G.; Cornuet, J.M. Bottleneck: A computer program for detecting recent reductions in the effective population size using allele frequency data. J. Hered. 1999, 90, 502–503. [Google Scholar] [CrossRef]
- Cornuet, J.-M.; Pudlo, P.; Veyssier, J.; Dehne-Garcia, A.; Gautier, M.; Leblois, R.; Marin, J.-M.; Estoup, A. DIYABC v2. 0: A software to make approximate Bayesian computation inferences about population history using single nucleotide polymorphism, DNA sequence and microsatellite data. Bioinformatics 2014, 30, 1187–1189. [Google Scholar] [CrossRef]
- Cabrera, A.A.; Palsbøll, P.J. Inferring past demographic changes from contemporary genetic data: A simulation-based evaluation of the ABC methods implemented in DIYABC. Mol. Ecol. Resour. 2017, 17, e94–e110. [Google Scholar] [CrossRef] [PubMed]
- Templeton, A.R.; Sing, C. A cladistic analysis of phenotypic associations with haplotypes inferred from restriction endonuclease mapping. IV. Nested analyses with cladogram uncertainty and recombination. Genetics 1993, 134, 659–669. [Google Scholar] [CrossRef] [PubMed]
- Billington, N.; Hebert, P. Mitochondrial DNA diversity in fishes and its implications for introductions. Can. J. Fish. Aquat. Sci. 1991, 48, 80–94. [Google Scholar] [CrossRef]
- Booy, G.; Hendriks, R.J.J.; Smulders, M.J.M.; Van Groenendael, J.M.; Vosman, B. Genetic diversity and the survival of populations. Plant Biol. 2000, 2, 379–395. [Google Scholar] [CrossRef]
- Searle, J.B. Phylogeography—The History and Formation of Species. Heredity 2000, 85, 201. [Google Scholar] [CrossRef]
- Hoelzel, A.; Hancock, J.; Dover, G. Evolution of the cetacean mitochondrial D-loop region. Mol. Biol. Evol. 1991, 8, 475–493. [Google Scholar]
- Sun, P.; Shi, Z.-h.; Yin, F.; Peng, S.-m. Genetic Variation Analysis of Mugil cephalus in China Sea Based on Mitochondrial COI Gene Sequences. Biochem. Genet. 2012, 50, 180–191. [Google Scholar] [CrossRef]
- Hua, X.; Wang, W.; Yin, W.; He, Q.; Jin, B.; Li, J.; Chen, J.; Fu, C. Phylogeographical analysis of an estuarine fish, Salanx ariakensis (Osmeridae: Salanginae) in the north-western Pacific. J. Fish Biol. 2009, 75, 354–367. [Google Scholar] [CrossRef]
- Yu, Z.N.; Kong, X.Y.; Guo, T.H.; Jiang, Y.-y.; Zhuang, Z.-m.; Jin, X.-s. Mitochondrial DNA sequence variation of Japanese anchovy Engraulis japonicus from the Yellow Sea and East China Sea. Fish. Sci. 2005, 71, 299. [Google Scholar] [CrossRef]
- Gu, S.; Yi, M.-R.; He, X.-B.; Lin, P.-S.; Liu, W.-H.; Luo, Z.-S.; Lin, H.-D.; Yan, Y.-R. Genetic diversity and population structure of cutlassfish (Lepturacanthus savala) along the coast of mainland China, as inferred by mitochondrial and microsatellite DNA markers. Reg. Stud. Mar. Sci. 2021, 43, 101702. [Google Scholar] [CrossRef]
- Zeng, L.; Cheng, Q.; Chen, X. Microsatellite analysis reveals the population structure and migration patterns of Scomber japonicus (Scombridae) with continuous distribution in the East and South China Seas. Biochem. Syst. Ecol. 2012, 42, 83–93. [Google Scholar] [CrossRef]
- Wang, X.; Weng, Z.; Yang, Y.; Hua, S.; Zhang, H.; Meng, Z. Genetic Evaluation of Black Sea Bream (Acanthopagrus schlegelii) Stock Enhancement in the South China Sea Based on Microsatellite DNA Markers. Fishes 2021, 6, 47. [Google Scholar] [CrossRef]
- Sun, D.-Q.; Shi, G.E.; Liu, X.-Z.; Wang, R.-X.; Xu, T.-J. Genetic diversity and population structure of the marbled rockfish, Sebastiscus marmoratus, revealed by SSR markers. J. Genet. 2013, 92, 21–24. [Google Scholar] [CrossRef]
- Falconer, D.S.; Mackay, T.F. Quantitative Genetics; Longman: London, UK, 1983. [Google Scholar]
- To, A.W.; De Mitcheson, Y.S. Shrinking baseline: The growth in juvenile fisheries, with the Hong Kong grouper fishery as a case study. Fish Fish. 2009, 10, 396–407. [Google Scholar] [CrossRef]
- Chen, B.; Luo, H.Z.; Fu, R.B. Biology and hatchery of Epinephelus awoara. Hebei Fish. 2006, 2, 29–31. [Google Scholar]
- To, W.L. The Biology, Fisheries of Groupers (Family: Serranidae) in Hong Kong and Adjacent Waters, and Implications for Management. Ph.D Thesis, University of Hong Kong, Hong Kong, 2009. [Google Scholar]
- Bjørnstad, O.N.; Grenfell, B.T. Noisy clockwork: Time series analysis of population fluctuations in animals. Science 2001, 293, 638–643. [Google Scholar] [CrossRef]
- Sun, J.; Cornelius, S.P.; Janssen, J.; Gray, K.A.; Motter, A.E. Regularity underlies erratic population abundances in marine ecosystems. J. R. Soc. Interface 2015, 12, 20150235. [Google Scholar] [CrossRef]
- Banks, S.C.; Cary, G.J.; Smith, A.L.; Davies, I.D.; Driscoll, D.A.; Gill, A.M.; Lindenmayer, D.B.; Peakall, R. How does ecological disturbance influence genetic diversity? Trends Ecol. Evol. 2013, 28, 670–679. [Google Scholar] [CrossRef]
- Romiguier, J.; Gayral, P.; Ballenghien, M.; Bernard, A.; Cahais, V.; Chenuil, A.; Chiari, Y.; Dernat, R.; Duret, L.; Faivre, N. Comparative population genomics in animals uncovers the determinants of genetic diversity. Nature 2014, 515, 261–263. [Google Scholar] [CrossRef]
- Ellegren, H.; Galtier, N. Determinants of genetic diversity. Nat. Rev. Genet. 2016, 17, 422–433. [Google Scholar] [CrossRef]
- Wright, S. Size of population and breeding structure in relation to evolution. Science 1938, 87, 430–431. [Google Scholar]
- Palumbi, S.R. Genetic divergence, reproductive isolation, and marine speciation. Annu. Rev. Ecol. Syst. 1994, 25, 547–572. [Google Scholar] [CrossRef]
- Shi, M.; Chen, C.; Xu, Q.; Lin, H.; Liu, G.; Wang, H.; Wang, F.; Yan, J. The role of Qiongzhou Strait in the seasonal variation of the South China Sea circulation. J. Phys. Oceanogr. 2002, 32, 103–121. [Google Scholar] [CrossRef]
- Chen, C.; Li, P.; Shi, M.; Zuo, J.; Chen, M.; Sun, H. Numerical study of the tides and residual currents in the Qiongzhou Strait. Chin. J. Oceanol. Limnol. 2009, 27, 931–942. [Google Scholar] [CrossRef]
- Qiu, B.; Imasato, N. A numerical study on the formation of the Kuroshio Counter Current and the Kuroshio Branch Current in the East China Sea. Cont. Shelf Res. 1990, 10, 165–184. [Google Scholar] [CrossRef]
- Zhao, D.; Li, Q.; Kong, L.; Yu, H. Cryptic diversity of marine gastropod Monodonta labio (Trochidae): Did the early Pleistocene glacial isolation and sea surface temperature gradient jointly drive diversification of sister species and/or subspecies in the Northwestern Pacific? Mar. Ecol. 2017, 38, e12443. [Google Scholar] [CrossRef]
- Wang, Z.-D.; Liao, J.; Huang, C.-Q.; Long, S.-S.; Zhang, S.; Guo, Y.-S.; Liu, L.; Liu, C.-W. Significant genetic differentiation of Gobiopterus lacustris, a newly recorded transparent goby in China. Mitochondrial DNA Part A 2018, 29, 785–791. [Google Scholar] [CrossRef]
- Peng, S.; Shi, Z.; Hou, J.; Wang, W.; Zhao, F.; Zhang, H. Genetic diversity of silver pomfret (Pampus argenteus) populations from the China Sea based on mitochondrial DNA control region sequences. Biochem. Syst. Ecol. 2009, 37, 626–632. [Google Scholar] [CrossRef]
- Voris, H.K. Maps of Pleistocene sea levels in Southeast Asia: Shorelines, river systems and time durations. J. Biogeogr. 2000, 27, 1153–1167. [Google Scholar] [CrossRef]
- Liu, J.X.; Gao, T.X.; Wu, S.F.; Zhang, Y.P. Pleistocene isolation in the Northwestern Pacific marginal seas and limited dispersal in a marine fish, Chelon haematocheilus (Temminck & Schlegel, 1845). Mol. Ecol. 2007, 16, 275–288. [Google Scholar]
- Liu, S.-Y.V.; Huang, I.-H.; Liu, M.-Y.; Lin, H.-D.; Wang, F.-Y.; Liao, T.-Y. Genetic stock structure of Terapon jarbua in Taiwanese waters. Mar. Coast. Fish. 2015, 7, 464–473. [Google Scholar] [CrossRef]
- Imbrie, J.; Boyle, E.; Clemens, S.; Duffy, A.; Howard, W.; Kukla, G.; Kutzbach, J.; Martinson, D.; McIntyre, A.; Mix, A. On the structure and origin of major glaciation cycles 1. Linear responses to Milankovitch forcing. Paleoceanography 1992, 7, 701–738. [Google Scholar] [CrossRef]
- Hewitt, G. The genetic legacy of the Quaternary ice ages. Nature 2000, 405, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.Q.; Gao, T.X.; Yanagimoto, T.; Sakurai, Y. Genetic population structure of Nibea albiflora in yellow sea and east china sea. Fish. Sci. 2008, 74, 544–552. [Google Scholar] [CrossRef]
- Neo, M.L.; Liu, L.-L.; Huang, D.; Soong, K. Thriving populations with low genetic diversity in giant clam species, Tridacna maxima and Tridacna noae, at Dongsha Atoll, South China Sea. Reg. Stud. Mar. Sci. 2018, 24, 278–287. [Google Scholar] [CrossRef]
- Yan, Y.-R.; Hsu, K.-C.; Yi, M.-R.; Li, B.; Wang, W.-K.; Kang, B.; Lin, H.-D. Cryptic diversity of the spotted scat Scatophagus argus (Perciformes: Scatophagidae) in the South China Sea: Pre-or post-production isolation. Mar. Freshw. Res. 2020, 71, 1640–1650. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sampling Site(Population Code) | N a | mtDNA | RyR3 | Microsatellite | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| h b | H c | θπ d | θω e | H b | H c | θπ d | θω e | Aa f | AR g | HO h | HE i | FIS j | HWE k | ||
| Ningde (MB) | 33 | 30 | 0.994 | 0.0044 | 0.0083 | 5 | 0.173 | 0.00032 | 0.0010 | 20.3 ± 1.248 | 14.71 | 0.836 ± 0.035 | 0.917 ± 0.007 | 0.103 | 0.156 |
| Xiamen (MN) | 36 | 27 | 0.971 | 0.0037 | 0.0061 | 5 | 0.366 | 0.00061 | 0.0013 | 18.8 ± 1.356 | 15.64 | 0.826 ± 0.036 | 0.907 ± 0.010 | 0.104 | 0.223 |
| Shenzhen (DYW) | 34 | 27 | 0.980 | 0.0033 | 0.0059 | 6 | 0.169 | 0.00031 | 0.0016 | 19.9 ± 1.676 | 15.80 | 0.858 ± 0.036 | 0.908 ± 0.010 | 0.070 | 0.419 |
| Weizhou (BBW) | 17 | 11 | 0.926 | 0.0034 | 0.0037 | 6 | 0.410 | 0.00084 | 0.0015 | 13.4 ± 0.806 | 15.46 | 0.876 ± 0.045 | 0.870 ± 0.045 | 0.023 | 0.079 |
| Mean | 82 | 0.968 | 0.0037 | 0.0060 | 0.280 | 0.00052 | 0.0013 | 18.1 ± 0.769 | 15.40 | 0.849 ± 0.019 | 0.900 ± 0.006 | ||||
| MB | MN | DYW | BBW | |
|---|---|---|---|---|
| MB | −0.0104 | −0.0055 | −0.0070 | |
| MN | 0.0090 *** | −0.0005 | −0.0203 | |
| DYW | 0.0039 | 0.0060 * | 0.0121 | |
| BBW | 0.0235 *** | 0.0180 *** | 0.0208 *** |
| Source of Variation | mtDNA | nuDNA | Microsatellite | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Variance Components | Percentage Variation | p | Variance Components | Percentage Variation | p | Variance Components | Percentage Variation | p | |
| 1. Scenario I: Four groups (MB) (MN) (DYW) (BBW) | |||||||||
| Among groups | −0.0244 | −0.57 | 0.6657 | 0.0022 | 1.37 | 0.0391 | 0.0453 | 0.97 | 0.4024 |
| Among individuals within populations | 4.3226 | 100.57 | 0.0000 | 0.1555 | 98.63 | 0.0000 | 0.3832 | 8.24 | 0.0000 |
| Within individuals | 4.2246 | 90.79 | 0.0000 | ||||||
| 2. Scenario II: Two groups (MB) (MN, DYW, BBW) [divided by the Taiwan Strait] | |||||||||
| Among groups | −0.0059 | −0.14 | 0.7429 | −0.0025 | −1.61 | 1.0000 | −0.0110 | −0.24 | 0.5063 |
| Among populations within groups | −0.0212 | −0.49 | 0.4565 | 0.0035 | 2.26 | 0.0342 | 0.0514 | 1.11 | 0.0000 |
| Among individuals within populations | 4.3226 | 100.63 | 0.6618 | 0.1555 | 99.35 | 0.0352 | 0.3832 | 8.24 | 0.0000 |
| Within individuals | 4.2246 | 90.89 | 0.0000 | ||||||
| 3. Scenario III: Two groups (MB, MN) (DYW, BBW) | |||||||||
| Among groups | −0.0179 | −0.42 | 0.6804 | −0.0020 | −1.27 | 1.0000 | −0.0171 | −0.37 | 1.0000 |
| Among populations within groups | −0.0125 | −0.29 | 0.4976 | 0.0035 | 2.22 | 0.0127 | 0.0567 | 1.22 | 0.0000 |
| Among individuals within populations | 4.3226 | 100.71 | 0.6383 | 0.1555 | 99.04 | 0.0264 | 0.3832 | 8.25 | 0.0000 |
| Within individuals | 4.2246 | 90.90 | 0.0000 | ||||||
| 4. Scenario IV: Two groups (MB, MN, DYW) (BBW) [divided by the Qiongzhou Strait] | |||||||||
| Among groups | −0.0049 | −0.11 | 0.5132 | 0.0066 | 4.07 | 0.2463 | 0.0635 | 1.35 | 0.2452 |
| Among populations within groups | −0.0228 | −0.53 | 0.6471 | 0.0000 | −0.01 | 0.3500 | 0.0240 | 0.51 | 0.0011 |
| Among individuals within populations | 4.3226 | 100.64 | 0.6540 | 0.1555 | 95.94 | 0.0274 | 0.3832 | 8.16 | 0.0000 |
| Within individuals | 4.2246 | 89.97 | 0.0000 | ||||||
| 5. Scenario V: Three groups (MB) (MN, DYW) (BBW) [divided by the Taiwan Strait and Qiongzhou Strait] | |||||||||
| Among groups | −0.0193 | −0.45 | 0.8348 | 0.0026 | 1.67 | 0.3636 | 0.0299 | 0.64 | 0.3277 |
| Among populations within groups | −0.0096 | −0.22 | 0.3803 | 0.0001 | 0.09 | 0.3558 | 0.0222 | 0.48 | 0.0233 |
| Among individuals within populations | 4.3226 | 100.67 | 0.6461 | 0.1555 | 98.24 | 0.0303 | 0.3832 | 8.22 | 0.0000 |
| Within individuals | 4.2246 | 90.66 | 0.0000 | ||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, K.; Lin, H.; Liu, R.; Ding, S. Genetic Structure and Demographic History of Yellow Grouper (Epinephelus awoara) from the Coast of Southeastern Mainland China, Inferred by Mitochondrial, Nuclear and Microsatellite DNA Markers. Diversity 2022, 14, 439. https://doi.org/10.3390/d14060439
Yang K, Lin H, Liu R, Ding S. Genetic Structure and Demographic History of Yellow Grouper (Epinephelus awoara) from the Coast of Southeastern Mainland China, Inferred by Mitochondrial, Nuclear and Microsatellite DNA Markers. Diversity. 2022; 14(6):439. https://doi.org/10.3390/d14060439
Chicago/Turabian StyleYang, Kuan, Hungdu Lin, Ruiqi Liu, and Shaoxiong Ding. 2022. "Genetic Structure and Demographic History of Yellow Grouper (Epinephelus awoara) from the Coast of Southeastern Mainland China, Inferred by Mitochondrial, Nuclear and Microsatellite DNA Markers" Diversity 14, no. 6: 439. https://doi.org/10.3390/d14060439
APA StyleYang, K., Lin, H., Liu, R., & Ding, S. (2022). Genetic Structure and Demographic History of Yellow Grouper (Epinephelus awoara) from the Coast of Southeastern Mainland China, Inferred by Mitochondrial, Nuclear and Microsatellite DNA Markers. Diversity, 14(6), 439. https://doi.org/10.3390/d14060439