Abstract
Mitochondrial metagenomics (MMG) using Illumina sequencers for mixed-species samples provides a promising tool for evolutionary and ecological studies using mitogenomes. However, the traditional assembly procedure is still computationally intensive and expensive. Here, a novel MMG pipeline was applied to different DNA extractions, one per species, and their sequence as a mixed sample for rapid mitogenome assembly is presented. Our method integrated a faster and more accurate read mapper for filtering non-mitochondrial reads. A seed-and-extend assembler for species-specific mitogenomes that detects ‘noisy species/sequences’ was also assessed. The MMG pipeline for each dataset was completed in a few hours on desktop PCs, maintaining high accuracy and completeness (COI divergence >10%), except for some very closely related taxa. Particularly for closely related species, the exclusion of ‘noisy reads’ (including chimera of non-targeted species) improved the target assembly. In addition, we observed that short barcodes used as references had almost identical detection power compared with mitogenomes but required greater sequencing depth. We tested our MMG pipeline on two real and one simulated dataset to validate its high efficiency in mixed-species sample mitogenome assembly.
1. Introduction
Understanding biodiversity, i.e., the variety and variability of life forms on Earth, has been recognized as a fundamental topic in evolutionary and ecological biology. Individual-based DNA techniques using Sanger sequencing can speed up species identification as complementary diagnoses for morphological taxonomy [1,2]. These studies usually analyze sequence divergence by amplifying nuclear and organellar gene regions, such as the standard Cytochrome C Oxidase Subunit I (COI) barcode for animals [1]. The successor metabarcoding can assess biodiversity from environmental DNA and bulk/mixed-species samples by amplicon next-generation sequencing (NGS) [3]. All of the above DNA-based approaches require PCR amplification and often suffer from some shortcomings, such as amplification difficulty across a broad taxonomic range [4], high requirements for samples (e.g., need fresh samples), and the experiment being complicated and time-consuming [5]. The recently emerged mitochondrial metagenomics (MMG) [6,7] can further unify the ecological and evolutionary understanding of biodiversity [8,9]. MMG has been applied in systematics and community phylogenetics by assembling high-copy mitochondrial contigs from low-coverage shotgun sequencing of specimen mixture [6,10,11,12,13,14].
Despite the great potential for evolutionary and ecological studies, MMG has not been considered perfect in the efficiency of mitogenome assembly using published pipelines. These assembly procedures are usually time-consuming and labor-intensive [8]: filter non-mitochondrial reads by BLAST [15] searches against a mitogenome database, assemble putative mitochondrial reads using multiple assemblers, annotate preliminary contigs, and combine overlapping contigs to generate longer consensus sequences [6,7]. DNA searches with BLASTn are not suitable for aligning very divergent sequences [16,17]. In contrast, Burrows Wheeler transformation (BWT)-based short read mappers, e.g., BWA [18], often have higher sensitivity in several orders of magnitude in less time [19], although they are also less efficient in coping with high sequence divergence. Compared to BWT-based mappers, hash-based ones, e.g., NextGenMap [20] and NOVOPlasty [21], can handle highly polymorphic reads and genomes in a shorter running time. Unfortunately, the disk and memory usage for hash-based assembly in NOVOPlasty can be substantial in a regular PC (i.e., PC with less than 16 G memory). Therefore, read mappers completely have the potential ability to replace BLAST for the read filtering with higher accuracy in much less time.
MMG assembly usually uses tools initially designed for whole-genome data rather than AT-rich mitogenome, e.g., IDBA-UD [22], Celera Assembler [23], SOAPdenovo [24], SOAPdenovo-Trans [25], and Newbler [26]. To maximize sequence contiguity, subsequent contig consensus requires additional tools and manual checks, such as Minimus [27], TGICL [28], and Geneious [29]. Two major categories of mitogenome-specific assemblers exist: (1) seed-free assembly software, such as MitoZ [30] and MEANGS [31], which can directly assemble mitogenomes from raw data without seed/bait sequences; (2) seed-based assembly software, such as MITObim [32] and NOVOPlasty, which can directly assemble mitogenomes with the user-provided seed/bait sequences for single-species samples. The MitoZ assembly is accurate; however, it is very time- and computing-resources-consuming [30]. The MEANGS is a seed-free de novo assembly software that applies tree-search to extend contigs from self-discovery seeds and assembles the mitogenome from NGS data [31]. Nonetheless, MitoZ and MEANGS were not suitable to extract the mitochondrial genome from mixed-species samples. The earlier version (v1.6) of MITObim provided a trial proofreading algorithm for metagenomic data. It was only tested in samples with a few (<6) species [32,33] but failed in more complex samples of typical MMG pools (unpublished data). Many rounds of iterations in MITObim are very time-consuming and hinder its further applications in metagenomic data. NOVOPlasty is faster and more accurate than other software, allowing for seeds from the same species or genus, even different genera, and families. A relaxed seed design is a benefit for single-species samples but becomes a shortcoming for complex samples.
This study aims to improve the MMG assembly efficiency by integrating faster, more accurate, and low-consumption bioinformatic tools. The short read mapper of NextGenMap is used to filter non-mitochondrial reads. The mitogenome-specific assembler NOVOPlasty was used for mitogenome assembly by restricting its seed compatibility. We tested the pipeline on both real and simulated raw sequencing datasets, covering both distantly and closely related species.
2. Materials and Methods
2.1. Data Generation
We tested the MMG assembly pipeline on two real and one simulated sequencing datasets that have reference mitogenome sequences (COI barcode also included) for each species. Dataset DS_A (SRA174290, 150 PE) included 49 divergent animal species (see in Supplementary Materials Table S1 and Figure S1a) [7]. Dataset DS_B [34] represented an extremely closely related-bee-species case: 48 individual raw sequencing data (100 PE) were combined into a mixture (Table S2); COI p-distance among species varied from 0.036 to 0.289 (mean 0.207) (Figure S1b). To better evaluate the performance of our pipeline in the presence of closely related species, we generated a dataset DS_C including 10 mosquito species, for which the interspecific p-distance was 0.015–0.166 (mean 0.109) (Figure S1c); the mitochondrial coverage was set as 200x to guarantee a sufficient amount with the ratio of mitochondrial to nuclear reads as 1% for each species (Table S3). The simulation of raw sequencing reads was generated using ART [35] with a read length of 150 bp, an insert fragment size of 300 bp (standard deviation of 30 bp), and a sequencing error of 0.1% (art_illumina-ss HSXn-i GENOME.fasta-p-l 150-f 200-m 300-s 30-na-qs 30-qs2 30-o SPECIES.mito.).
2.2. Mitogenome Assembly
The workflow of MMG mitogenome assembly is shown in Figure 1. All analyses were executed in the CentOS 7 operating system on an AMD RYZEN 1700X CPU (8 cores/16 threads) and 32 G memory PC. Commands for merging all the forward or reverse reads, and the script of MMG are available on GitHub (https://github.com/xtmtd/MMG, accessed on 14 March 2022). Raw sequencing data were mapped to a mitogenome database using NextGenMap v0.5.5 with an identity threshold of 0.3. The sequences of mitogenome database were downloaded from NCBI Reference Sequence Database (RefSeq, 15 July, 2018): 1632 (117 Arachnida, eight Asteroidea, 11 Branchiopoda, six Danio, 1490 Insecta) for DS_A, 16 (Apoidea) for DS_B, 3 (one Aedes, one Anopheles, one Culex) for DS_C. Candidate paired mitochondrial reads, in which at least one of the paired ones can be mapped, were extracted with SAMtools v1.7 [36]. Mitogenomes were assembled with a loop script that included the NOVOPlasty v4.3.1 assembler while storing hash locally to speed up the assembly for possible multiple runs. Each mitogenome assembly requires a seed or bait sequence, a short mitochondrial fragment used for the initial assembly. In practice, seeds can be generated using barcoding for each species or metabarcoding for multiplexed species pool (DNA libraries with a unique identifier for each species). COI of 658 bp (standard ‘barcode’) and 313 bp (‘mini-barcode’, named mini-COI here) were selected as seeds and extracted from the reference in this study. Assembled contig(s) of each species were aligned to seeds of all species with VSEARCH v2.7.1 (--usearch_global --blast6out --maxaccepts 0 --maxrejects 0 --id 0.2) [37]; a contig of identity greater than 0.95 (i.e., highly matching seed sequences, query cover 100%) was assigned to the species from NOVOPlasty outputs, and then, mitogenomes were assembled.
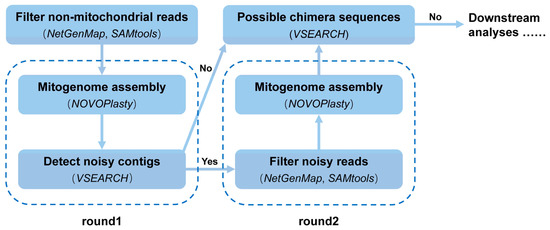
Figure 1.
Workflow of the MMG assembly. Bioinformatic tools used in each step are marked as italic.
Assembled contig(s) from different NOVOPlasty procedures may be identical for closely related species due to the relaxed seed compatibility. With the species-specific seed sequence (COI here), NOVOPlasty may assemble mitochondrial contigs belonging to other species present in the same sample. This means that the presence of ‘noisy reads’ of non-targeted ‘noisy species’ in the raw sequencing data obstructs the target assembly. The exclusion of these ‘noisy reads’ would alleviate this difficulty. These ‘noisy species’ were identified when the assembled contigs matched the seeds of non-targeted species revealed by VSEARCH results. Noisy reads were filtered against ‘noisy mitochondrial contigs’ assembled in the previous assembly round using NextGenMap, SAMtools with an identity value of 0.99. The resulting reads were used to assemble mitogenomes again for the species that failed in the previous assembly round. Then, the steps of the above noise detection-filtering-assembly procedure were repeated (usually one–two rounds) until no more noisy reads were discovered. Specific mitogenome reference sequences were drawn from Tang et al. [7,34] or NCBI public ones. Due to the great assembly difficulty in dataset DS_B, a second seed of 497-bp ND5 was used for assembly, and species with contigs shorter than 5000 bp were re-assembled in the filtering rounds.
Assembled contigs could be heterospecific contigs (chimera), although their partial regions well matched the seeds. We examined the chimera as follows: contigs were divided into multiple 200 bp fragments, and a VSEARCH search against all assembled mitogenomes was performed for each fragment. A contig having best matches (identity ≥ 97%) with two or more reference genomes was treated as a chimera. Those ‘noisy’ parts within the chimera were filtered and newer assemblies were generated following the above noise-filtering steps.
The final assembly was finished until no noisy regions including chimeras were detected. The assembly quality was measured relative to the reference using genome coverage (percentage of the reference genome) and accuracy (percentage of correctly assembled nucleotides for aligned regions). Only one round of filtering noisy reads was performed for most species. For the closely related species, two rounds of filtering noisy reads were needed. The complete process of mitochondrial genome assembly was executed following the custom script.
Three preconditions were used to assemble mitogenomes using the MMG pipeline: (a) having seeds in advance; (b) knowing that species inside the sample are not too similar, as to avoid cross assembly; and (c) ensuring that the DNA of each species is mixed in the right quantity.
3. Results
A total of 45.8, 130.8, and 3.1 Gbp of raw PE reads were generated for datasets DS_A, DS_B, and DS_C, respectively. After the removal of non-mitochondrial reads, 6.88%, 5.12%, and 1.32% of raw reads were filtered out for the subsequent assembly. Detailed assembly results and statistics can be found in Tables S1–S3. Each NOVOPlasty assembly was finished in less than ten minutes depending on the amount of input candidate mitochondrial reads. In the initial assembly, the use of longer COI as seed sequences generated more noisy assembled reads than shorter mini-COI seeds for DS_A (3 vs. 0) and DS_C (4 vs. 2). Thus, COI was not selected as seeds in DS_B due to the great number of closely related species. For the dataset DS_B, the assembly of six species failed to produce any sequences in the first round of assembly with mini-COI seeds, and three assemblies generated chimera although correct seed sequences were included in a chimera (Table S2). Chimeras of 14,974 bp assembled from two species of DS_C are highly similar to both reference sequences (Anopheles gambiae and A. merus), which had a 0.015 p-distance for COI. After one or two rounds of filtering noisy reads, all target assemblies were correctly recovered except for two species in DS_C.
The mitogenome assembly lengths were 14,361.2 ± 3182.7 (3,876–17,633) bp, 8510.2 ± 5241.2 (630–17,211) bp, and 15,582.4 ± 445.5 (15,377–16,673) bp for DS_A, DS_B, and DS_C, respectively, corresponding to the genome coverage (%) relative to the references 95.1 ± 20.1, 58.2 ± 34.2, and 100 ± 0.1 (Figure 2). Ten and seven mitogenomes of DS_A and DS_B, respectively, were slightly longer than the reference. For the dataset DS_B, assembly with ND5 seeds produced contigs (1044–7857 bp) not overlapping with mini-COI results for ten species (Table S2). The mitogenome accuracy for aligned regions was 99.9% ± 0.6%, 99.7% ± 0.4%, 99.6% ± 0.7% for DS_A, DS_B, and DS_C, respectively. Twenty-one mitogenome sequences in DS_A were circularized while 20 were in the reference.
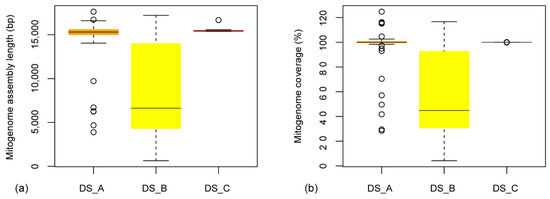
Figure 2.
(a) Mitogenome assembly length and (b) coverage for three datasets.
4. Discussion
Our novel MMG pipeline greatly accelerated the assembly by efficient improvements in removing non-mitochondrial reads, particularly in simplifying the workflow and reducing running time (the run times were within 12 h, 20 h, and 8 h for datasets DS_A, DS_B, and DS_C, respectively). Assembly contiguity and accuracy can be comparable with the references (Figure 2) for both distantly and closely related taxa. Detection and removal of noisy reads, including chimera, further guarantee the correctness of target sequences.
Seed or bait sequences for each species are prerequisites for assembly using NOVOPlasty. Seeds can be generated from standard barcoding or metabarcoding, with the latter pooling multiple amplification products to further reduce the cost [3]. In addition, the 313 bp mini-COI, which is the most frequently used marker in metabarcoding [38], outperforms the standard 658 bp-COI when serving as seeds for assembly, resulting in fewer incorrect assemblies (Tables S1 and S3). A single seed can produce nearly complete mitogenomes when sequencing coverage is enough [8], as exemplified in both DS_A and DS_C. Additional seeds (ND5 in DS_B, which was acquired from the same genome) may help to generate more contigs for one species [7], particularly when mitochondrial coverage is limited (often less than 50x) and sequencing read length is short (e.g., 100 PE in DS_B).
Although the seed-and-extend algorithm in NOVOPlasty greatly reduces the chimera for mixed-species samples, it is possible to fail for very closely related species (Tables S2 and S3). Chimera detection and removal in our pipeline eliminated most errors but still failed in two mosquito species with 1.5% COI divergence (Table S3). According to our tests, we recommend that the accuracy and completeness of COI divergence >10% are necessary (see the details in Appendix A).
Our assessments of simulated datasets (Appendix A) indicate that species, even at the very low sequencing coverage (0.1x), can be assembled mitogenomes in bulk samples. Short barcodes as the reference have similar detection powers but require at least an order of magnitude greater in sequencing depth (much higher cost) than whole mitogenomes. Rapid decreases in sequencing costs, e.g., library preparation of CNY 20 plus CNY 4/Gbp per sample on the BGI or NovaSeq 6000 platforms (price from BerryGenomics, China, 1 March, 2022), allows MMG to be applied in a wider scope for ecological and evolutionary studies [8,9] (e.g., the origin and phylodiversity of insects [39,40], relative species abundance estimation [39]).
Supplementary Materials
The following supporting information can be downloaded at https://www.mdpi.com/article/10.3390/d14050317/s1, Figure S1: The COI p-distance among species and phylogenetic relationships of datasets DS_A (a), DS_B (b), and DS_C (c); Table S1: Species information, mitogenome reference sequences, and assembly results for dataset DS_A. The yellow cells represent the chimeras, and the species name of the same sequence are written in brackets; Table S2: Species information, mitogenome reference sequences, and assembly results for dataset DS_B. The yellow cells represent the chimeras, and the species name and the length of the same sequence are written in brackets. The grey cells represent the fail to assemble; Table S3: Species information, mitogenome and genome reference sequences, and assembly results for dataset DS_C. The yellow cells represent the chimeras, and the species name of the same sequence are written in the brackets; Table S4: Assessment of the accuracy of MMG seed-based approach at varying mitochondrial coverages against mitogenome, COI, and mini-COI references, respectively.
Author Contributions
Conceptualization, J.D. and F.Z.; Data curation, S.D., J.D., J.W. and F.Z.; Funding acquisition, J.W. and F.Z.; Methodology, S.D., J.D. and F.Z.; Writing—original draft, F.Z.; Writing—review and editing, S.D. and N.N.G. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (No. 31970434) to F.Z. and the Project of Biological Resources Survey in Wuyishan National Park to J.W.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Commands of merging all the forward or reverse reads, and the script of MMG (MMG.sh) used in this study are available at GitHub (https://github.com/xtmtd/MMG, accessed on 14 March 2022). Details and script usage are provided also on the same webpage.
Acknowledgments
We are grateful to the two anonymous reviewers for their constructive comments and suggestions that substantially improved the quality of this manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
Appendix A
To test the accuracy of read-based approach (species detection rate), we simulated seven 10-mosquito-species datasets of sequencing data with the mitochondrial coverage respectively at 0.1, 1, 10, 50, 200, 500, 1000× and the ratio of mitochondrial to nuclear reads as 1% for each species using ART. Species abundance was detected against mitogenome (AT-rich regions excluded), barcode (658 bp-COI) and mini-barcode (313 bp-COI) references using a custom loop script, which employed HS-BLASTN v.0.0.5 [41] and BLAST+ v2.7.1 [42] as aligner and identity of 0.99. Required minimum sequencing amount for a MMG bulk sample was assessed assuming 100 species, average 1% ratio of mitochondrial/genomic DNA, and mitochondrial coverage of 0.1× in the sample.
Species detection was tested on datasets only including closely-related species (Figure S1c). Read numbers were accurately estimated (Pearson R2 = 0.97, p < 0.001), using R package (version 4.1.2; https://www.r-project.org/, accessed on 14 March 2022), against mitogenome reference at 0.1–1000× sequencing coverage except for Anopheles gambiae and A. merus (p-distance of 0.015) (Figure A1a, Table S4). Species detection failed at the coverage of 0.1× against COI reference (Figure A1b) and 0.1× and 1× against mini-COI reference (Figure A1c), but estimated read numbers with other coverages were highly correlated with the number of simulated mitochondrial reads. The required minimum sequencing amount per MMG bulk sample was 15,000 bp*(0.1x)/1%*(100 species) = 15 Mbp, i.e., sequencing amount of 1 Gbp per bulk sample is usually sufficient to detect species richness and abundance. It is helping us to better understand and practice the minimum sequencing coverage when mix the samples by species detection, and saving experiment costs or computing resources maximally.
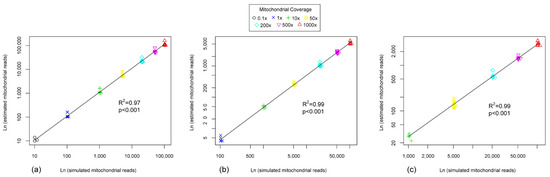
Figure A1.
Assessing species abundance (read numbers) at varying mitochondrial coverage against mitogenome (a), COI (b), and mini-COI (c) references, respectively.
References
- Hebert, P.D.N.; Cywinska, A.; Ball, S.L.; DeWaard, J.R. Biological identifications through DNA barcodes. Proceeding R. Soc. B Biol. Sci. 2003, 270, 313–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tautz, D.; Arctander, P.; Minelli, A.; Thomas, R.H.; Vogler, A.P. A plea for DNA taxonomy. Trends Ecol. Evol. 2003, 18, 70–74. [Google Scholar] [CrossRef]
- Taberlet, P.; Coissac, E.; Pompanon, F.; Brochmann, C.; Willerslev, E. Towards next-generation biodiversity assessment using DNA metabarcoding. Mol. Ecol. 2012, 21, 2045–2050. [Google Scholar] [CrossRef] [PubMed]
- Deagle, B.E.; Jarman, S.N.; Coissac, E.; Pompanon, F.; Taberlet, P. DNA metabarcoding and the cytochrome c oxidase subunit I marker: Not a perfect match. Biol. Lett. 2014, 10, 20140562. [Google Scholar] [CrossRef] [Green Version]
- Tamura, K.; Aotsuka, T. Rapid isolation method of animal mitochondrial DNA by the alkaline lysis procedure. Biochem. Genet. 1988, 26, 815–819. [Google Scholar] [CrossRef]
- Crampton-Platt, A.; Timmermans, M.J.T.N.; Gimmel, M.L.; Kutty, S.N.; Cockerill, T.D.; Khen, C.V.; Vogler, A.P. Soup to Tree: The Phylogeny of Beetles Inferred by Mitochondrial Metagenomics of a Bornean Rainforest Sample. Mol. Biol. Evol. 2015, 32, 2302–2316. [Google Scholar] [CrossRef]
- Tang, M.; Tan, M.; Meng, G.; Yang, S.; Su, X.; Liu, S.; Song, W.; Li, Y.; Wu, Q.; Zhang, A.; et al. Multiplex sequencing of pooled mitochondrial genomes-a crucial step toward biodiversity analysis using mito-metagenomics. Nucleic Acids Res. 2014, 42, e166. [Google Scholar] [CrossRef] [Green Version]
- Crampton-Platt, A.; Yu, D.W.; Zhou, X.; Vogler, A.P. Mitochondrial metagenomics: Letting the genes out of the bottle. GigaScience 2016, 5, 15. [Google Scholar] [CrossRef] [Green Version]
- Arribas, P.; Andújar, C.; Hopkins, K.; Shepherd, M.; Vogler, A.P. Metabarcoding and mitochondrial metagenomics of endogean arthropods to unveil the mesofauna of the soil. Methods Ecol. Evol. 2016, 9, 1071–1081. [Google Scholar] [CrossRef]
- Gillett, C.P.D.T.; Crampton-Platt, A.; Timmermans, M.J.T.N.; Jordal, B.H.; Emerson, B.C.; Vogler, A.P. Bulk de novo mitogenome assembly from pooled total DNA elucidates the phylogeny of weevils (Coleoptera: Curculionoidea). Mol. Biol. Evol. 2014, 31, 2223–2237. [Google Scholar] [CrossRef] [Green Version]
- Gómez-Rodríguez, C.; Crampton-Platt, A.; Timmermans, M.J.; Baselga, A.; Vogler, A.P. Validating the power of mitochondrial metagenomics for community ecology and phylogenetics of complex assemblages. Methods Ecol. Evol. 2015, 6, 883–894. [Google Scholar] [CrossRef]
- Choo, L.Q.; Crampton-Platt, A.; Vogler, A.P. Shotgun mitogenomics across body size classes in a local assemblage of tropical Diptera: Phylogeny, species diversity and mitochondrial abundance spectrum. Mol. Ecol. 2017, 26, 5086–5098. [Google Scholar] [CrossRef] [PubMed]
- Cicconardi, F.; Borges, P.A.V.; Strasberg, D.; Oromí, P.; López, H.; Pérez-Delgado, A.J.; Casquet, J.; Caujapé-Castells, J.; Fernández-Palacios, J.M.; Thébaud, C.; et al. MtDNA metagenomics reveals large-scale invasion of belowground arthropod communities by introduced species. Mol. Ecol. 2017, 26, 3104–3115. [Google Scholar] [CrossRef] [Green Version]
- Nie, R.-E.; Breeschoten, T.; Timmermans, M.J.T.N.; Nadein, K.; Xue, H.J.; Bai, M.; Huang, Y.; Yang, X.K.; Vogler, A.P. The phylogeny of Galerucinae (Coleoptera: Chrysomelidae) and the performance of mitochondrial genomes in phylogenetic inference compared to nuclear rRNA genes. Cladistics 2018, 34, 113–130. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Pearson, W.R. BLAST and FASTA Similarity Searching for Multiple Sequence Alignment. Methods Mol. Biol. 2014, 1079, 75–101. [Google Scholar]
- Rahman, F.; Hassan, M.; Kryshchenko, A.; Dubchak, I.; Tatarinova, T.V.; Alexandrov, N. benchNGS: An approach to benchmark short reads alignment tools. arXiv 2015, arXiv:1504.06659. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Li, R.; Li, Y.; Kristiansen, K.; Wang, J. SOAP: Short oligonucleotide alignment program. Bioinformatics 2008, 24, 713–714. [Google Scholar] [CrossRef] [Green Version]
- Sedlazeck, F.J.; Rescheneder, P.; von Haeseler, A. NextGenMap: Fast and accurate read mapping in highly polymorphic genomes. Bioinformatics 2013, 29, 2790–2791. [Google Scholar] [CrossRef] [Green Version]
- Dierckxsens, N.; Mardulyn, P.; Smits, G. NOVOPlasty: De novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 2017, 45, e18. [Google Scholar] [PubMed] [Green Version]
- Peng, Y.; Leung, H.C.; Yiu, S.M.; Chin, F.Y. IDBA-UD: A de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 2012, 28, 1420–1428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myers, E.W.; Sutton, G.G.; Delcher, A.L.; Dew, I.M.; Fasulo, D.P.; Flanigan, M.J.; Kravitz, S.A.; Mobarry, C.M.; Reinert, K.H.; Remington, K.A.; et al. A whole-genome assembly of Drosophila. Science 2000, 287, 2196–2204. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Zhu, H.; Ruan, J.; Qian, W.; Fang, X.; Shi, Z.; Li, Y.; Li, S.; Shan, G.; Kristiansen, K.; et al. De novo assembly of human genomes with massively parallel short read sequencing. Genome Res. 2010, 20, 265–272. [Google Scholar] [CrossRef] [Green Version]
- Xie, Y.; Wu, G.; Tang, J.; Luo, R.; Patterson, J.; Liu, S.; Huang, W.; He, G.; Gu, S.; Li, S.; et al. SOAPdenovo-Trans: De novo transcriptome assembly with short RNA-Seq reads. Bioinformatics 2014, 30, 1660–1666. [Google Scholar] [CrossRef] [Green Version]
- Margulies, M.; Egholm, M.; Altman, W.E.; Attiya, S.; Bader, J.S.; Bemben, L.A.; Berka, J.; Braverman, M.S.; Chen, Y.J.; Chen, Z.; et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature 2005, 437, 376–380. [Google Scholar] [CrossRef]
- Sommer, D.D.; Delcher, A.L.; Salzberg, S.L.; Pop, M. Minimus: A fast, lightweight genome assembler. BMC Bioinform. 2007, 8, 64. [Google Scholar] [CrossRef] [Green Version]
- Pertea, G.; Huang, X.; Liang, F.; Antonescu, V.; Sultana, R.; Karamycheva, S.; Lee, Y.; White, J.; Cheung, F.; Parvizi, B.; et al. TIGR Gene Indices clustering tools (TGICL): A software system for fast clustering of large EST datasets. Bioinformatics 2003, 19, 651–652. [Google Scholar] [CrossRef] [Green Version]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Meng, G.; Li, Y.; Yang, C.; Liu, S. MitoZ: A Toolkit for Animal Mitochondrial Genome Assembly, Annotation and Visualization. Nucleic Acids Res. 2019, 47, e63. [Google Scholar] [CrossRef]
- Song, M.-H.; Yan, C.; Li, J.-T. MEANGS: An efficient seed-free tool for de novo assembling animal mitochondrial genome using whole genome NGS data. Bioinformatics 2021, 23, bbab538. [Google Scholar] [CrossRef] [PubMed]
- Hahn, C.; Bachmann, L.; Chevreux, B. Reconstructing mitochondrial genomes directly from genomic next-generation sequencing read-a baiting and iterative mapping approach. Nucleic Acids Res. 2013, 41, e129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Timbo, R.V.; Togawa, R.C.; Costa, M.M.; Andow, D.A.; Paula, D.P. Mitogenome sequence accuracy using different elucidation methods. PLoS ONE 2017, 12, e0179971. [Google Scholar]
- Tang, M.; Hardman, C.J.; Ji, Y.; Meng, G.; Liu, S.; Tan, M.; Yang, S.; Moss, E.D.; Wang, J.; Yang, C.; et al. High-throughput monitoring of wild bee diversity and abundance via mitogenomics. Methods Ecol. Evol. 2015, 6, 1034–1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, W.; Li, L.; Myers, J.R.; Marth, G.T. ART: A next-generation sequencing read simulator. Bioinformatics 2012, 28, 593–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools 1000 Genome Project Data Processing Subgroup. Bioinformatics 2012, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef]
- Leray, M.; Yang, J.Y.; Meyer, C.P.; Mills, S.C.; Agudelo, N.; Ranwez, V.; Boehm, J.T.; Machida, R.J. A new versatile primer set targeting a short fragment of the mitochondrial COI region for metabarcoding metazoan diversity: Application for characterizing coral reef fish gut contents. Front. Zool. 2013, 10, 34. [Google Scholar] [CrossRef] [Green Version]
- Garrido-Sanz, L.; Senar, M.À.; Piñol, J. Relative species abundance estimation in artificial mixtures of insects using mito-metagenomics and a correction factor for the mitochondrial DNA copy number. Mol. Ecol. Resour. 2021, 22, 153–167. [Google Scholar] [CrossRef]
- Arribas, P.; Andújar, C.; Moraza, M.L.; Linard, B.; Emerson, B.; Vogler, A.P. Mitochondrial Metagenomics Reveals the Ancient Origin and Phylodiversity of Soil Mites and Provides a Phylogeny of the Acari. Mol. Biol. Evol. 2019, 37, 683–694. [Google Scholar] [CrossRef]
- Chen, Y.; Ye, W.; Zhang, Y.; Xu, Y. High speed BLASTN: An accelerated MegaBLAST search tool. Nucleic Acids Res. 2015, 43, 7762–7768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).