
De Novo Metagenomic Analysis of Microbial Community Contributing in Lignocellulose Degradation in Humus Samples Harvested from Cuc Phuong Tropical Forest in Vietnam


 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
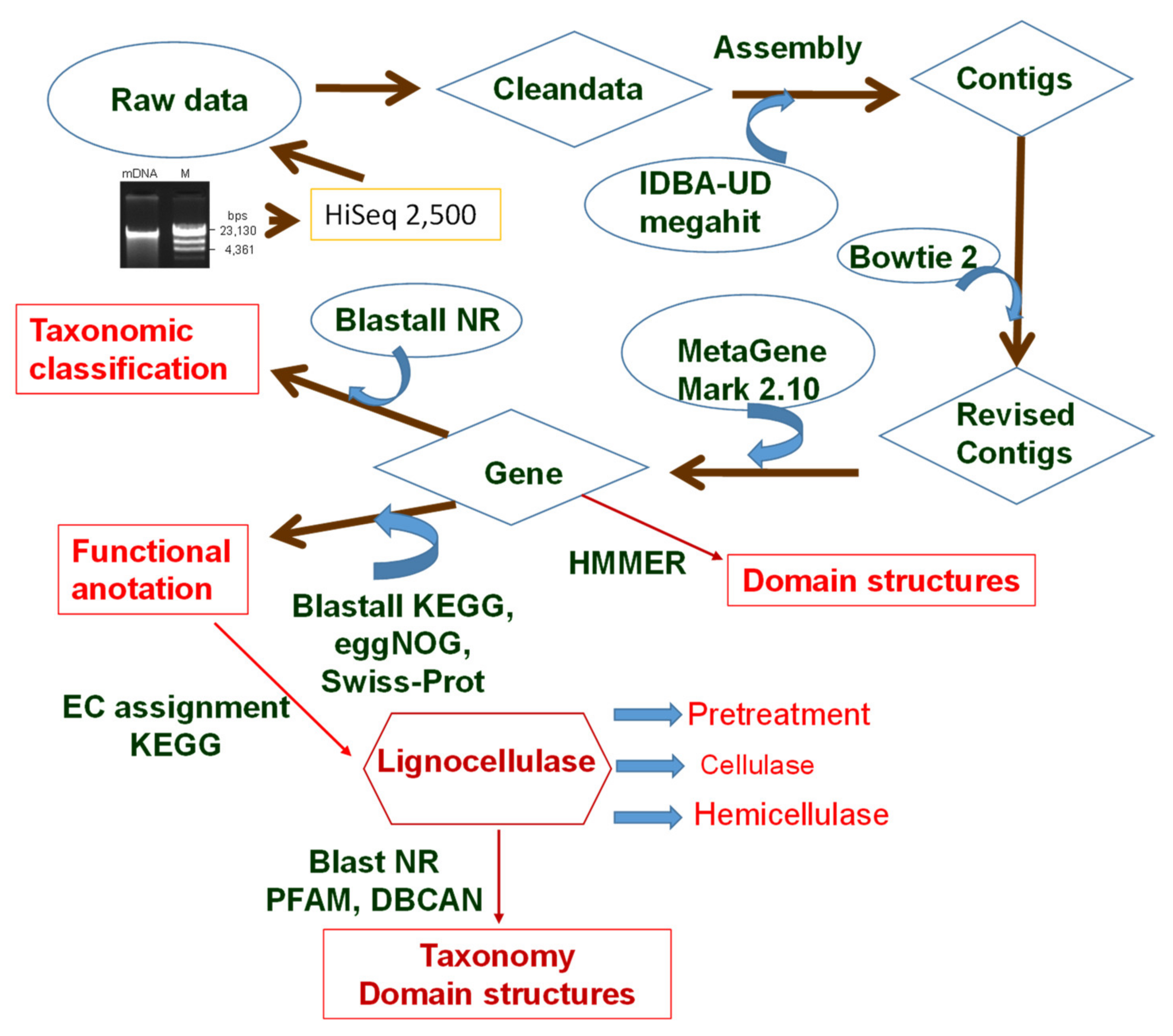
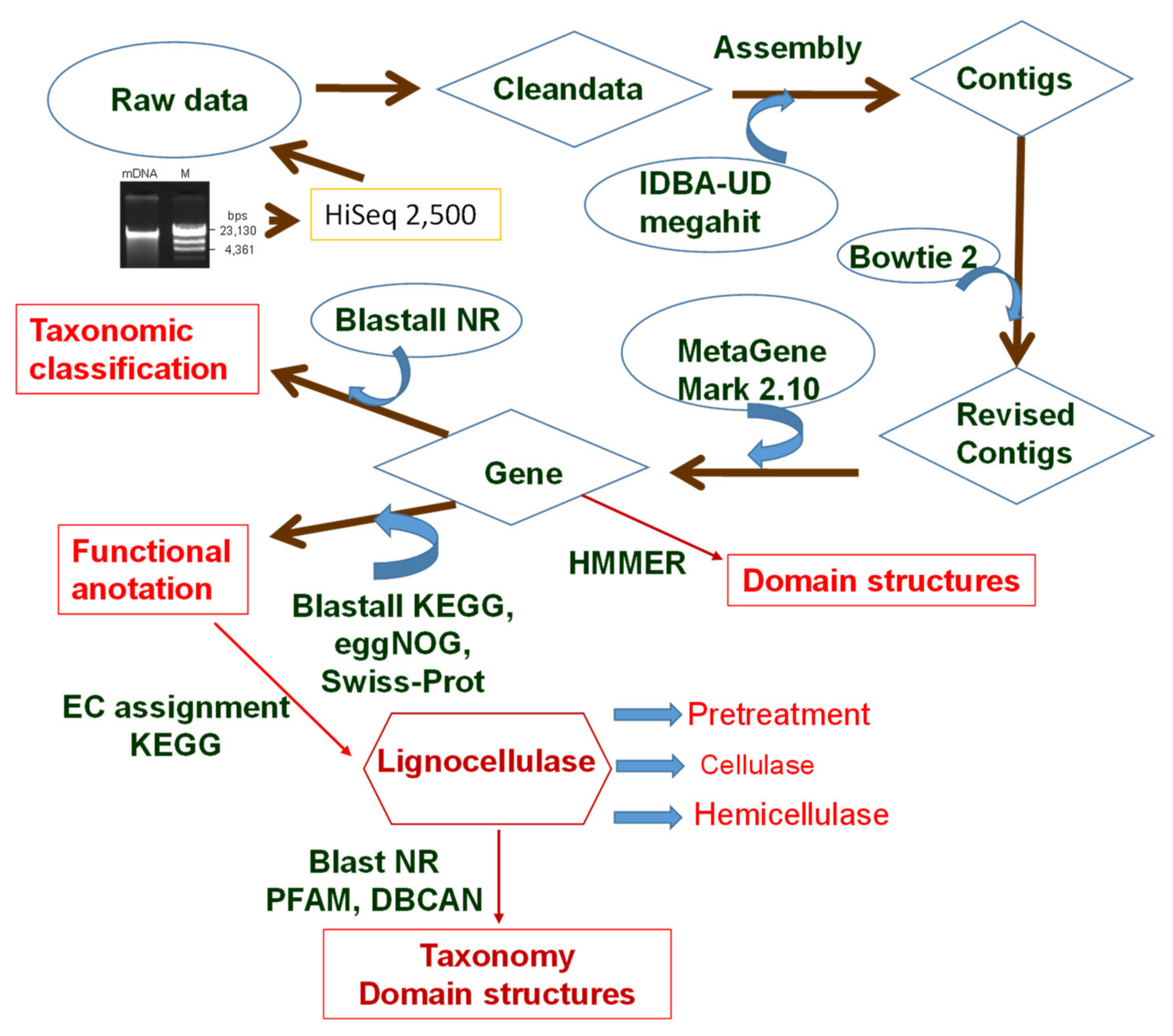
2.1. Sampling and Extraction of Metagenomics DNA
2.2. Metagenomic Sequencing
2.3. Taxonomic Assignment
2.4. Functional Annotation
2.5. Mining Genes Encoding Lignocellulose-Degrading Enzymes
3. Results
3.1. Metagenome Sequencing of Cuc Phuong Tropical Forest Soil
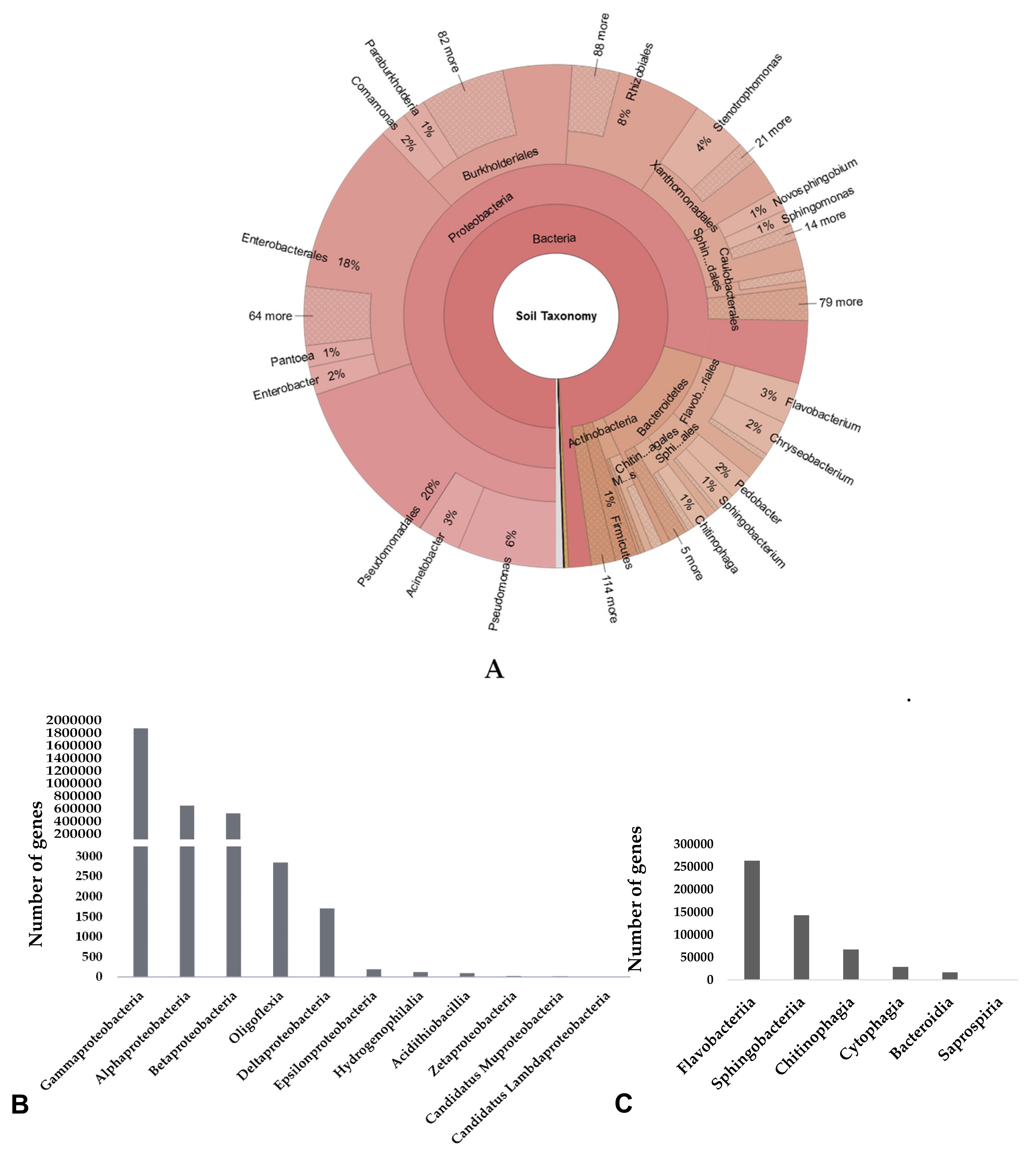
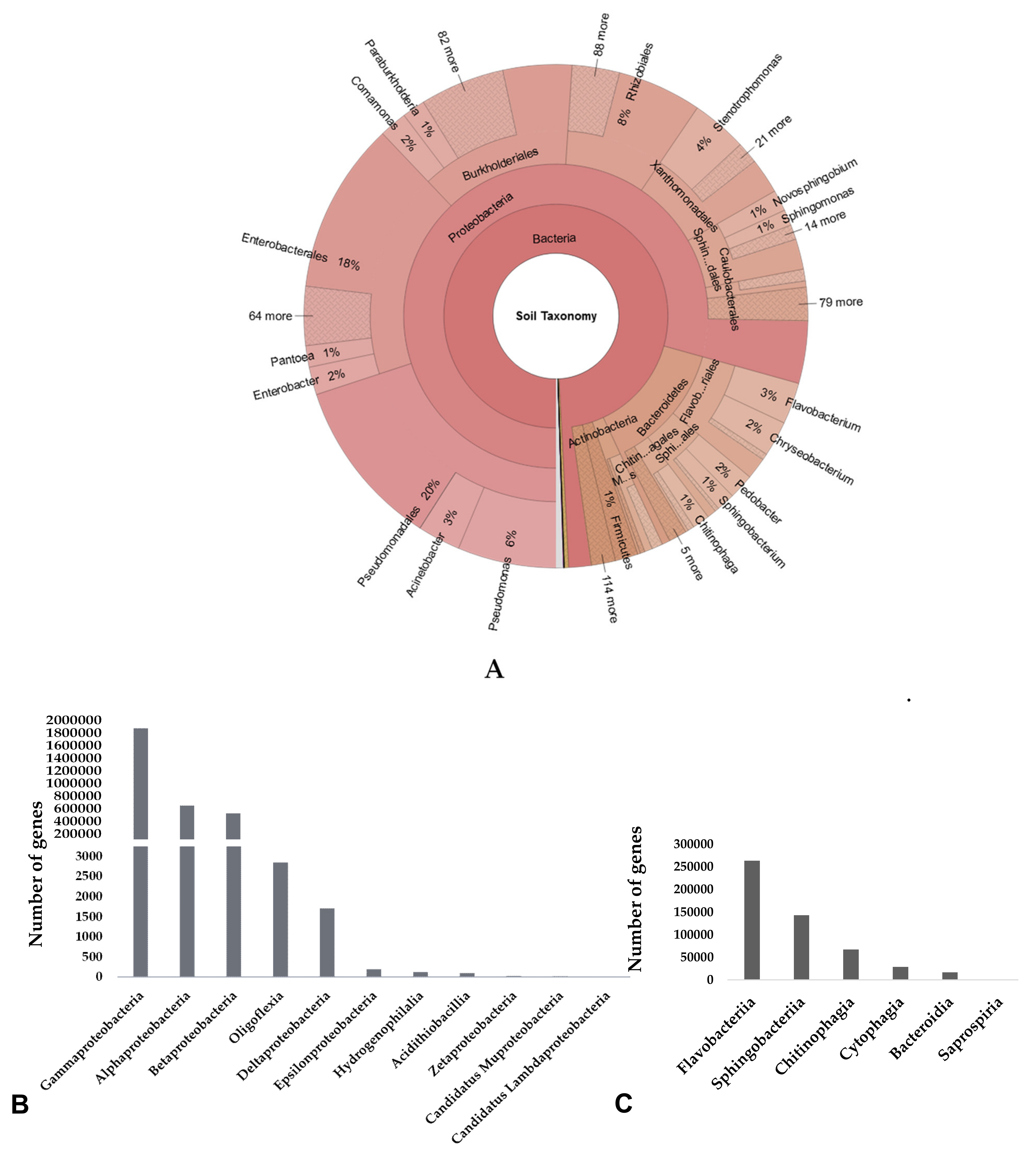
3.2. Taxonomic Composition of Microbial Community in Cuc Phuong’s Soil
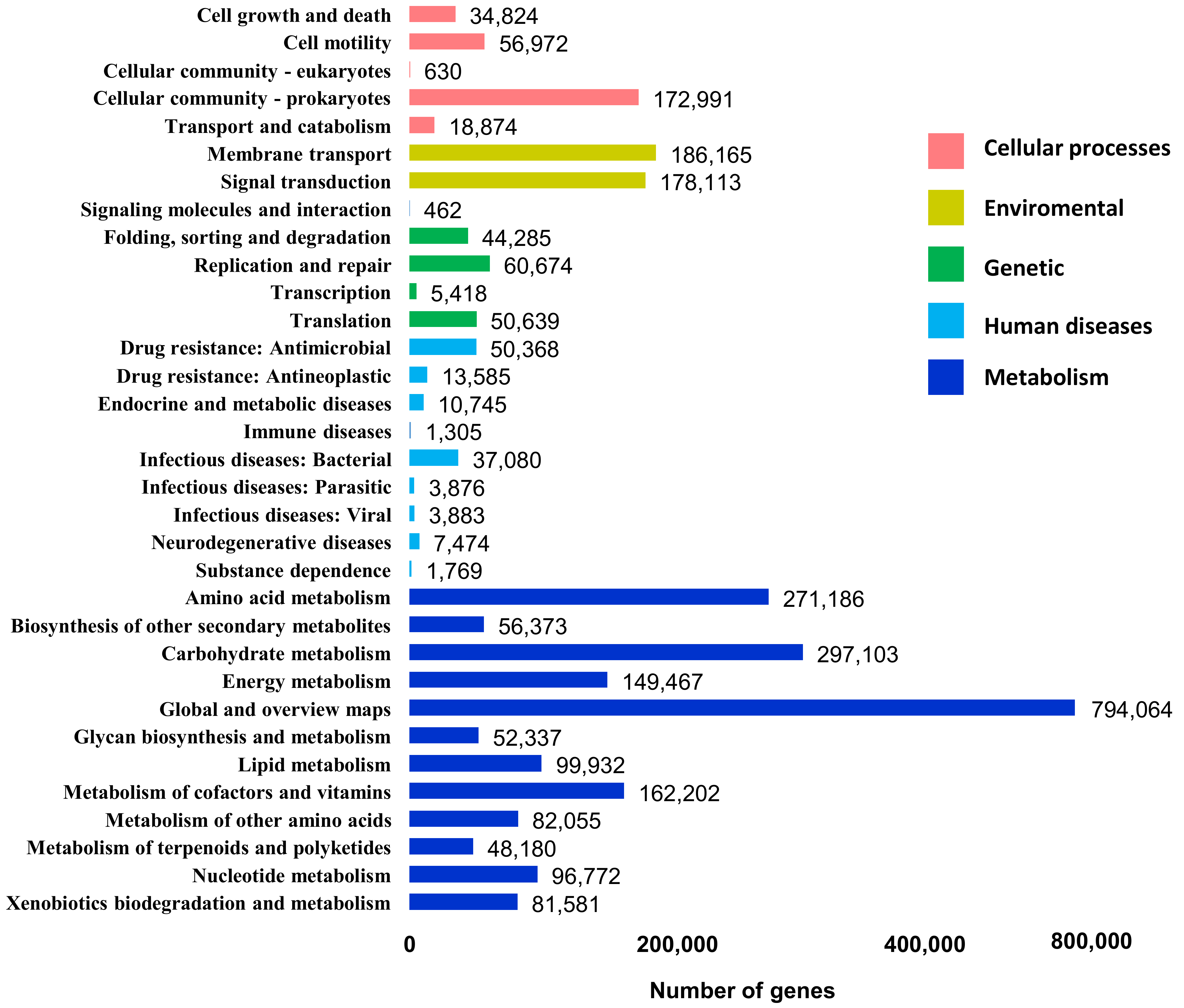
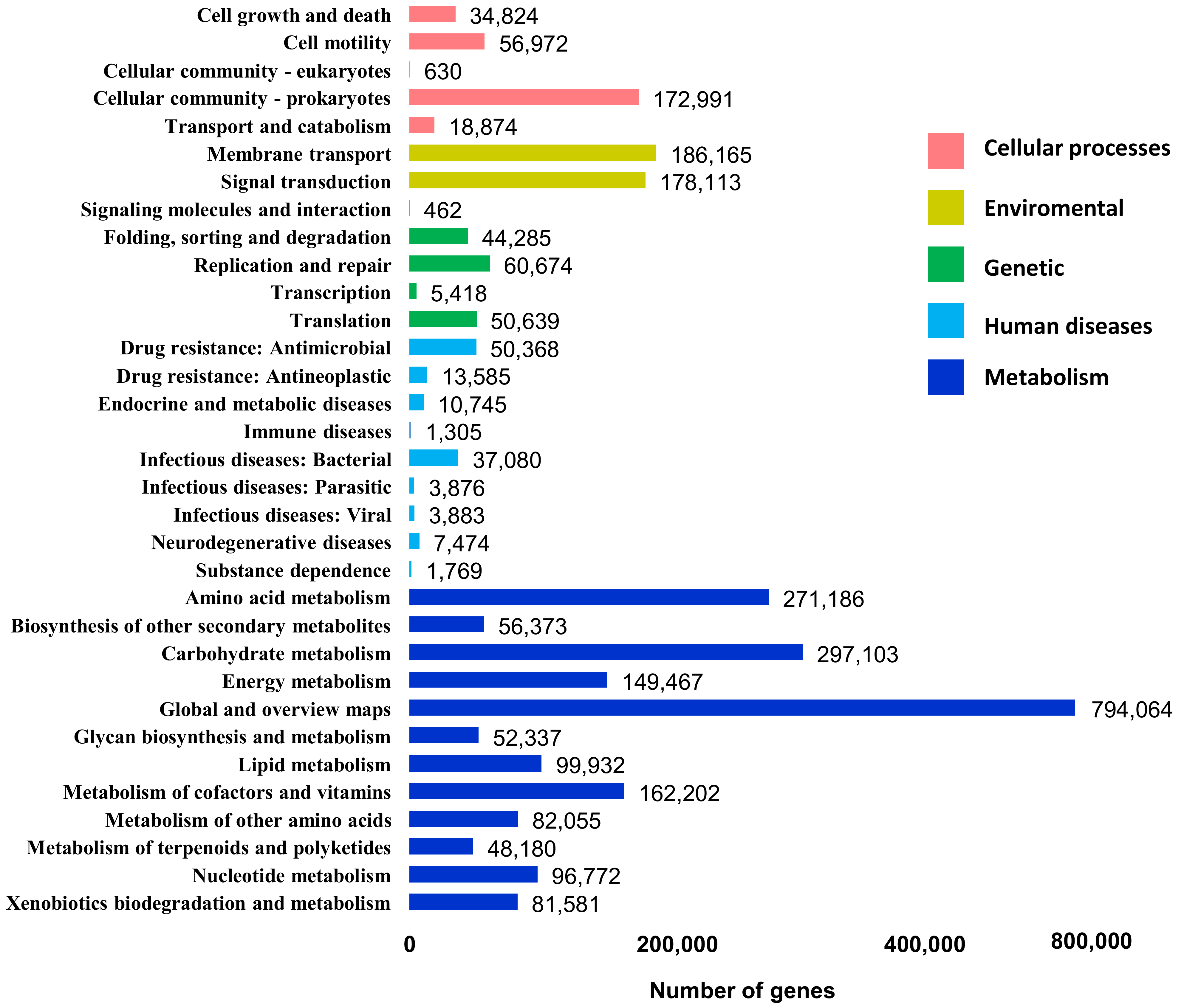
3.3. Functional Profile of DNA Metagenome from Cuc Phuong’s Humus
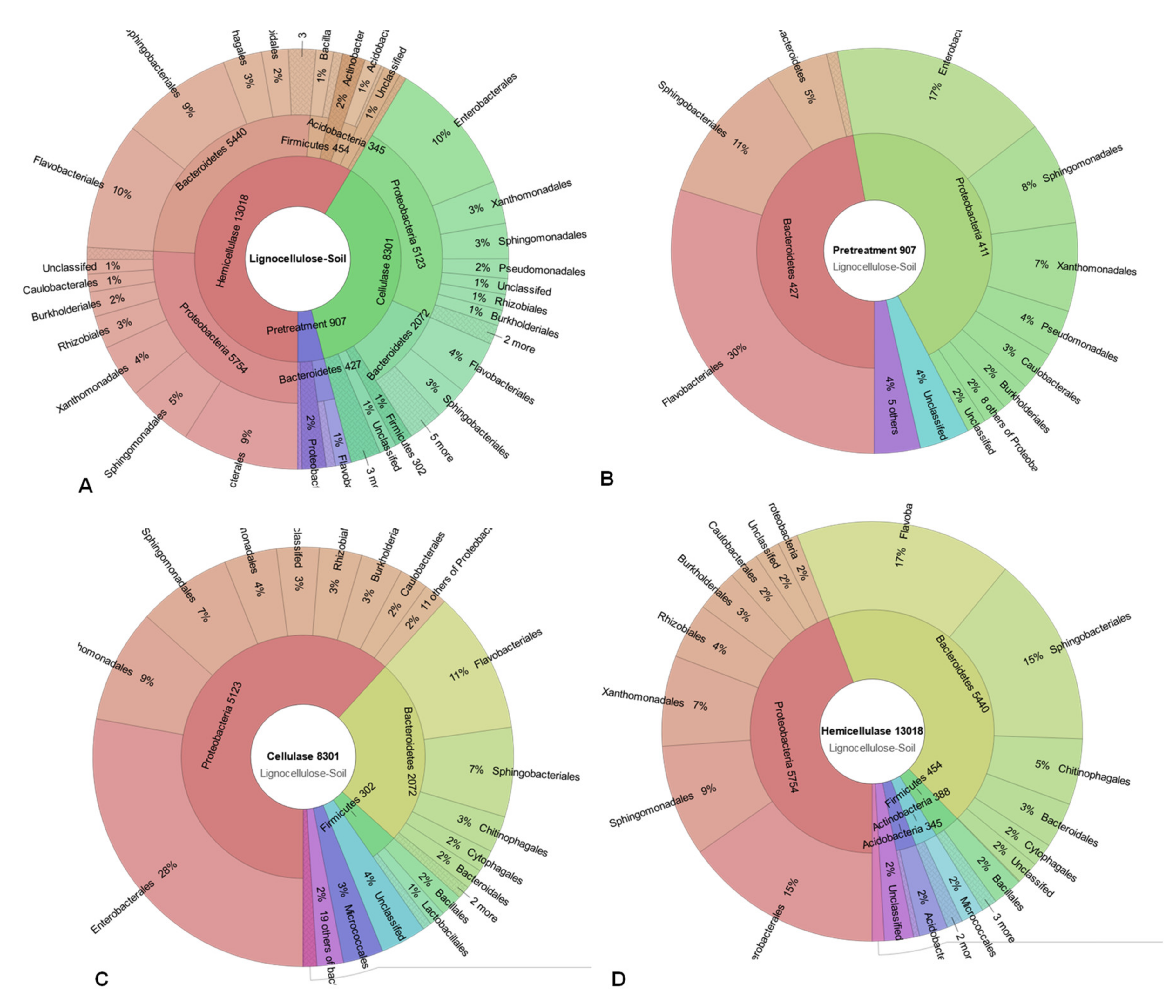
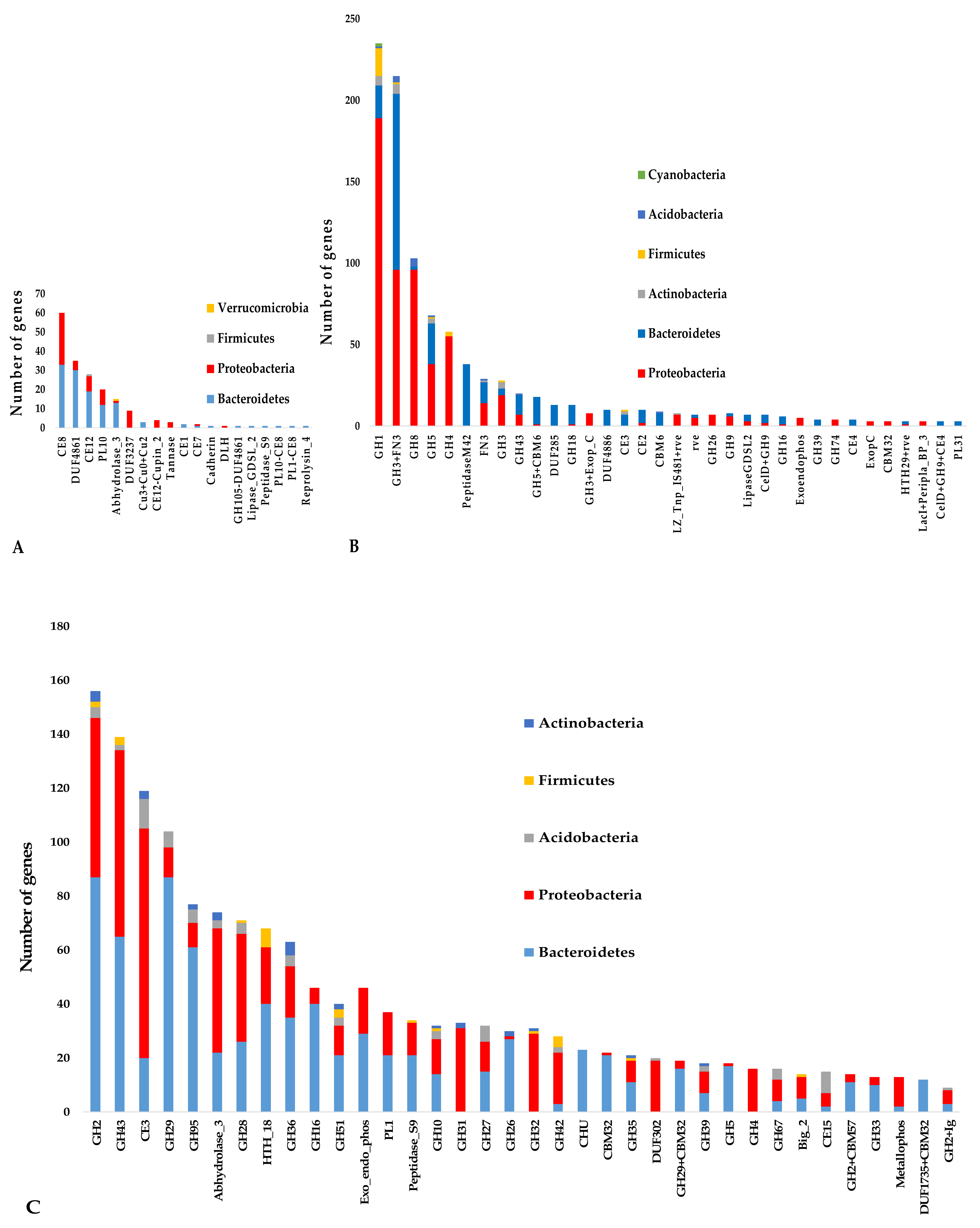
3.4. Putative Lignocellulose-Degrading Protein Encoding Genes
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sharma, H.K.; Xu, C.; Qin, W. Biological Pretreatment of Lignocellulosic Biomass for Biofuels and Bioproducts: An Overview. Waste Biomass Valoriz. 2019, 10, 235–251. [Google Scholar] [CrossRef]
- Bhatia, L.; Sharma, A.; Bachheti, R.K.; Chandel, A.K. Lignocellulose derived functional oligosaccharides: Production, properties, and health benefits. Prep. Biochem. Biotechnol. 2019, 49, 744–758. [Google Scholar] [CrossRef] [PubMed]
- Carriquiry, M.A.; Du, X.; Timilsina, G.R. Second generation biofuels: Economics and policies. Energy Policy 2011, 39, 4222–4234. [Google Scholar] [CrossRef] [Green Version]
- Himmel, M.E.; Ding, S.Y.; Johnson, D.K.; Adney, W.S.; Nimlos, M.R.; Brady, J.W.; Foust, T.D. Biomass recalcitrance: Engineering plants and enzymes for biofuels production. Science 2007, 315, 804–807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ragauskas, A.J.; Williams, C.K.; Davison, B.H.; Britovsek, G.; Cairney, J.; Eckert, C.A.; Frederick, W.J.; Hallett, J.P.; Leak, D.J.; Liotta, C.L.; et al. The path forward for biofuels and biomaterials. Science 2006, 311, 484–489. [Google Scholar] [CrossRef] [Green Version]
- Ravindran, R.; Hassan, S.S.; Williams, G.A.; Jaiswal, A.K. A review on bioconversion of agro-industrial wastes to industrially important enzymes. Bioengineering 2018, 5, 93. [Google Scholar] [CrossRef] [Green Version]
- Achinivu, E.C. Protic ionic liquids for lignin extraction—A lignin characterization study. Int. J. Mol. Sci. 2018, 19, 428. [Google Scholar] [CrossRef] [Green Version]
- Kumari, D.; Singh, R. Pretreatment of lignocellulosic wastes for biofuel production: A critical review. Renew. Sustain. Energy Rev. 2018, 90, 877–891. [Google Scholar] [CrossRef]
- Sharma, A.; Kumar, R.; Rathi, M.; Bhatia, D.; Malik, D.K. Lignocellulose biodegradation: An advance technology for sustainable environment. Biosci. Biotechnol. Res. Commun. 2018, 11, 634–637. [Google Scholar] [CrossRef]
- Van den Burg, B. Extremophiles as a source for novel enzymes. Curr. Opin. Microbiol. 2003, 6, 213–218. [Google Scholar] [CrossRef]
- Ventorino, V.; Aliberti, A.; Faraco, V.; Robertiello, A.; Giacobbe, S.; Ercolini, D.; Amore, A.; Fagnano, M.; Pepe, O. Exploring the microbiota dynamics related to vegetable biomasses degradation and study of lignocellulose-degrading bacteria for industrial biotechnological application. Sci. Rep. 2015, 5, 8161. [Google Scholar] [CrossRef] [PubMed]
- López-Mondéjar, R.; Algora, C.; Baldrian, P. Lignocellulolytic systems of soil bacteria: A vast and diverse toolbox for biotechnological conversion processes. Biotechnol. Adv. 2019, 37, 107374. [Google Scholar] [CrossRef] [PubMed]
- Lladó, S.; López-Mondéjar, R.; Baldrian, P. Forest Soil Bacteria: Diversity, Involvement in Ecosystem Processes, and Response to Global Change. Microbiol. Mol. Biol. Rev. 2017, 81, e00063-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rappé, M.S.; Giovannoni, S.J. The Uncultured Microbial Majority. Annu. Rev. Microbiol. 2003, 57, 369–394. [Google Scholar] [CrossRef] [Green Version]
- Duan, C.J.; Feng, J.X. Mining metagenomes for novel cellulase genes. Biotechnol. Lett. 2010, 32, 1765–1775. [Google Scholar] [CrossRef]
- Thompson, C.E.; Beys-da-Silva, W.O.; Santi, L.; Berger, M.; Vainstein, M.H.; Guimarães, J.A.; Vasconcelos, A.T.R. A potential source for cellulolytic enzyme discovery and environmental aspects revealed through metagenomics of Brazilian mangroves. AMB Express 2013, 3, 65. [Google Scholar] [CrossRef] [Green Version]
- Alteio, L.V.; Schulz, F.; Seshadri, R.; Varghese, N.; Rodriguez-Reillo, W.; Ryan, E.; Goudeau, D.; Eichorst, S.A.; Malmstrom, R.R.; Bowers, R.M.; et al. Complementary Metagenomic Approaches Improve Reconstruction of Microbial Diversity in a Forest Soil. mSystems 2020, 5, e00768-19. [Google Scholar] [CrossRef] [Green Version]
- Baldrian, P.; Kolaiřík, M.; Štursová, M.; Kopecký, J.; Valášková, V.; Větrovský, T.; Žifčáková, L.; Šnajdr, J.; Rídl, J.; Vlček, Č.; et al. Active and total microbial communities in forest soil are largely different and highly stratified during decomposition. ISME J. 2012, 6, 248–258. [Google Scholar] [CrossRef] [Green Version]
- Lopes, A.R.; Manaia, C.M.; Nunes, O.C. Bacterial community variations in an alfalfa-rice rotation system revealed by 16S rRNA gene 454-pyrosequencing. FEMS Microbiol. Ecol. 2014, 87, 650–663. [Google Scholar] [CrossRef] [Green Version]
- Schreiter, S.; Ding, G.C.; Heuer, H.; Neumann, G.; Sandmann, M.; Grosch, R.; Kropf, S.; Smalla, K. Effect of the soil type on the microbiome in the rhizosphere of field-grown lettuce. Front. Microbiol. 2014, 5, 144. [Google Scholar] [CrossRef]
- Turlapati, S.A.; Minocha, R.; Bhiravarasa, P.S.; Tisa, L.S.; Thomas, W.K.; Minocha, S.C. Chronic N-amended soils exhibit an altered bacterial community structure in Harvard Forest, MA, USA. FEMS Microbiol. Ecol. 2013, 83, 478–493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, Y.; Leung, H.C.M.; Yiu, S.M.; Chin, F.Y.L. IDBA-UD: A de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 2012, 28, 1420–1428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, D.; Liu, C.M.; Luo, R.; Sadakane, K.; Lam, T.W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, W.; Lomsadze, A.; Borodovsky, M. Ab initio gene identification in metagenomic sequences. Nucleic Acids Res. 2010, 38, e132. [Google Scholar] [CrossRef] [Green Version]
- Huson, D.H.; Auch, A.F.; Qi, J.; Schuster, S.C. MEGAN analysis of metagenomic data. Genome Res. 2007, 17, 377–386. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T.; et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008, 36, D480–D484. [Google Scholar] [CrossRef]
- Powell, S.; Szklarczyk, D.; Trachana, K.; Roth, A.; Kuhn, M.; Muller, J.; Arnold, R.; Rattei, T.; Letunic, I.; Doerks, T.; et al. eggNOG v3.0: Orthologous groups covering 1133 organisms at 41 different taxonomic ranges. Nucleic Acids Res. 2012, 40, D284–D289. [Google Scholar] [CrossRef]
- Mistry, J.; Finn, R.D.; Eddy, S.R.; Bateman, A.; Punta, M. Challenges in homology search: HMMER3 and convergent evolution of coiled-coil regions. Nucleic Acids Res. 2013, 41, e121. [Google Scholar] [CrossRef] [Green Version]
- Mistry, J.; Chuguransky, S.; Williams, L.; Qureshi, M.; Salazar, G.A.; Sonnhammer, E.L.L.; Tosatto, S.C.E.; Paladin, L.; Raj, S.; Richardson, L.J.; et al. Pfam: The protein families database in 2021. Nucleic Acids Res. 2021, 49, D412–D419. [Google Scholar] [CrossRef]
- Li, J.G.; Shen, M.C.; Hou, J.F.; Li, L.; Wu, J.X.; Dong, Y.H. Effect of different levels of nitrogen on rhizosphere bacterial community structure in intensive monoculture of greenhouse lettuce. Sci. Rep. 2016, 6, 25305. [Google Scholar] [CrossRef] [PubMed]
- Feng, G.; Xie, T.; Wang, X.; Bai, J.; Tang, L.; Zhao, H.; Wei, W.; Wang, M.; Zhao, Y. Metagenomic analysis of microbial community and function involved in cd-contaminated soil. BMC Microbiol. 2018, 18, 11. [Google Scholar] [CrossRef] [PubMed]
- Rousk, J.; Bååth, E.; Brookes, P.C.; Lauber, C.L.; Lozupone, C.; Caporaso, J.G.; Knight, R.; Fierer, N. Soil bacterial and fungal communities across a pH gradient in an arable soil. ISME J. 2010, 4, 1340–1351. [Google Scholar] [CrossRef]
- Lauber, C.L.; Hamady, M.; Knight, R.; Fierer, N. Pyrosequencing-based assessment of soil pH as a predictor of soil bacterial community structure at the continental scale. Appl. Environ. Microbiol. 2009, 75, 5111–5120. [Google Scholar] [CrossRef] [Green Version]
- Cho, S.J.; Kim, M.H.; Lee, Y.O. Effect of pH on soil bacterial diversity. J. Ecol. Environ. 2016, 40, 10. [Google Scholar] [CrossRef] [Green Version]
- Dimitriu, P.A.; Grayston, S.J. Relationship between soil properties and patterns of bacterial β-diversity across reclaimed and natural boreal forest soils. Microb. Ecol. 2010, 59, 563–573. [Google Scholar] [CrossRef] [PubMed]
- Fierer, N. Embracing the unknown: Disentangling the complexities of the soil microbiome. Nat. Rev. Microbiol. 2017, 15, 579–590. [Google Scholar] [CrossRef]
- Berlemont, R.; Martiny, A.C. Genomic potential for polysaccharide deconstruction in bacteria. Appl. Environ. Microbiol. 2015, 81, 1513–1519. [Google Scholar] [CrossRef] [Green Version]
- Lombard, V.; Golaconda Ramulu, H.; Drula, E.; Coutinho, P.M.; Henrissat, B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2014, 42, D490–D495. [Google Scholar] [CrossRef] [Green Version]
- Tveit, A.; Schwacke, R.; Svenning, M.M.; Urich, T. Organic carbon transformations in high-Arctic peat soils: Key functions and microorganisms. ISME J. 2013, 7, 299–311. [Google Scholar] [CrossRef] [Green Version]
- Alessi, A.M.; Bird, S.M.; Bennett, J.P.; Oates, N.C.; Li, Y.; Dowle, A.A.; Polikarpov, I.; Young, J.P.W.; McQueen-Mason, S.J.; Bruce, N.C. Revealing the insoluble metasecretome of lignocellulose-degrading microbial communities. Sci. Rep. 2017, 7, 2356. [Google Scholar] [CrossRef] [PubMed]
- Jiménez, D.J.; Chaves-Moreno, D.; Van Elsas, J.D. Unveiling the metabolic potential of two soil-derived microbial consortia selected on wheat straw. Sci. Rep. 2015, 5, 13845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berlemont, R.; Allison, S.D.; Weihe, C.; Lu, Y.; Brodie, E.L.; Martiny, J.B.H.; Martiny, A.C. Cellulolytic potential under environmental changes in microbial communities from grassland litter. Front. Microbiol. 2014, 5, 639. [Google Scholar] [CrossRef] [PubMed]
- Himmel, M.E.; Xu, Q.; Luo, Y.; Ding, S.Y.; Lamed, R.; Bayer, E.A. Microbial enzyme systems for biomass conversion: Emerging paradigms. Biofuels 2010, 1, 323–341. [Google Scholar] [CrossRef] [Green Version]
- Koeck, D.E.; Pechtl, A.; Zverlov, V.V.; Schwarz, W.H. Genomics of cellulolytic bacteria. Curr. Opin. Biotechnol. 2014, 29, 171–183. [Google Scholar] [CrossRef] [PubMed]
- López-Mondéjar, R.; Zühlke, D.; Větrovský, T.; Becher, D.; Riedel, K.; Baldrian, P. Decoding the complete arsenal for cellulose and hemicellulose deconstruction in the highly efficient cellulose decomposer Paenibacillus O199. Biotechnol. Biofuels 2016, 9, 104. [Google Scholar] [CrossRef] [Green Version]
- Sukharnikov, L.O.; Cantwell, B.J.; Podar, M.; Zhulin, I.B. Cellulases: Ambiguous nonhomologous enzymes in a genomic perspective. Trends Biotechnol. 2011, 29, 473–479. [Google Scholar] [CrossRef] [Green Version]
- Do, T.H.; Dao, T.K.; Nguyen, K.H.V.; Le, N.G.; Nguyen, T.M.P.; Le, T.L.; Phung, T.N.; van Straalen, N.M.; Roelofs, D.; Truong, N.H. Metagenomic analysis of bacterial community structure and diversity of lignocellulolytic bacteria in Vietnamese native goat rumen. Asian-Australas. J. Anim. Sci. 2018, 31, 738–747. [Google Scholar] [CrossRef]
- Baldrian, P. Fungal laccases-occurrence and properties. FEMS Microbiol. Rev. 2006, 30, 215–242. [Google Scholar] [CrossRef] [Green Version]
- Odriozola, I.; Abrego, N.; Tláskal, V.; Zrůstová, P.; Morais, D.; Větrovský, T.; Ovaskainen, O.; Baldrian, P. Fungal Communities Are Important Determinants of Bacterial Community Composition in Deadwood. mSystems 2021, 6, e01017-20. [Google Scholar] [CrossRef]
- Do, T.H.; Nguyen, T.T.; Nguyen, T.N.; Le, Q.G.; Nguyen, C.; Kimura, K.; Truong, N.H. Mining biomass-degrading genes through Illumina-based de novo sequencing and metagenomic analysis of free-living bacteria in the gut of the lower termite Coptotermes gestroi harvested in Vietnam. J. Biosci. Bioeng. 2014, 118, 665–671. [Google Scholar] [CrossRef] [PubMed]
- DeAngelis, K.M.; Gladden, J.M.; Allgaier, M.; D’haeseleer, P.; Fortney, J.L.; Reddy, A.; Hugenholtz, P.; Singer, S.W.; Vander Gheynst, J.S.; Silver, W.L.; et al. Strategies for enhancing the effectiveness of metagenomic-based enzyme discovery in lignocellulolytic microbial communities. Bioenergy Res. 2010, 3, 146–158. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-Couto, S. Industrial and environmental applications of white-rot fungi. Mycosphere 2017, 8, 456–466. [Google Scholar] [CrossRef]
- Wesenberg, D.; Kyriakides, I.; Agathos, S.N. White-rot fungi and their enzymes for the treatment of industrial dye effluents. Biotechnol. Adv. 2003, 22, 161–187. [Google Scholar] [CrossRef]
- Ruiz-Dueñas, F.J.; Martínez, Á.T. Microbial degradation of lignin: How a bulky recalcitrant polymer is efficiently recycled in nature and how we can take advantage of this. Microb. Biotechnol. 2009, 2, 164–177. [Google Scholar] [CrossRef] [Green Version]
- Kirk, T.K.; Farrell, R.L. Enzymatic “combustion”: The microbial degradation of lignin. Annu. Rev. Microbiol. 1987, 41, 465–505. [Google Scholar] [CrossRef] [PubMed]
- Allgaier, M.; Reddy, A.; Park, J.I.; Ivanova, N.; D’Haeseleer, P.; Lowry, S.; Sapra, R.; Hazen, T.C.; Simmons, B.A.; Vandergheynst, J.S.; et al. Targeted discovery of glycoside hydrolases from a switchgrass-adapted compost community. PLoS ONE 2010, 5, e8812. [Google Scholar] [CrossRef]
- Žifčáková, L.; Větrovský, T.; Lombard, V.; Henrissat, B.; Howe, A.; Baldrian, P. Feed in summer, rest in winter: Microbial carbon utilization in forest topsoil. Microbiome 2017, 5, 122. [Google Scholar] [CrossRef] [Green Version]
- Štursová, M.; Žifčáková, L.; Leigh, M.B.; Burgess, R.; Baldrian, P. Cellulose utilization in forest litter and soil: Identification of bacterial and fungal decomposers. FEMS Microbiol. Ecol. 2012, 80, 735–746. [Google Scholar] [CrossRef]
- Kataeva, I.A.; Seidel, R.D.; Shah, A.; West, L.T.; Li, X.L.; Ljungdahl, L.G. The fibronectin type 3-like repeat from the Clostridium thermocellum cellobiohydrolase CbHa promotes hydrolysis of cellulose by modifying its surface. Appl. Environ. Microbiol. 2002, 68, 4292–4300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, S.Y.; Xu, Q.; Crowley, M.; Zeng, Y.; Nimlos, M.; Lamed, R.; Bayer, E.A.; Himmel, M.E. A biophysical perspective on the cellulosome: New opportunities for biomass conversion. Curr. Opin. Biotechnol. 2008, 19, 218–227. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, K.H.V.; Dao, T.K.; Nguyen, H.D.; Nguyen, K.H.; Nguyen, T.Q.; Nguyen, T.T.; Nguyen, T.M.P.; Truong, N.H.; Do, T.H. Some characters of bacterial cellulases in goats’ rumen elucidated by metagenomic DNA analysis and the role of fibronectin 3 module for endoglucanase function. Asian-Australas. J. Anim. Sci. 2021, 34, 867–879. [Google Scholar] [CrossRef] [PubMed]
- Wilson, D.B. Three microbial strategies for plant cell wall degradation. Ann. N. Y. Acad. Sci. 2008, 1125, 289–297. [Google Scholar] [CrossRef]
- Do, T.H.; Le, N.G.; Dao, T.K.; Nguyen, T.M.P.; Le, T.L.; Luu, H.L.; Nguyen, K.H.V.; Nguyen, V.L.; Le, L.A.; Phung, T.N.; et al. Metagenomic insights into lignocellulose-degrading genes through Illuminabased de novo sequencing of the microbiome in vietnamese native goats’ rumen. J. Gen. Appl. Microbiol. 2018, 64, 108–116. [Google Scholar] [CrossRef] [Green Version]
- Sweeney, M.D.; Xu, F. Biomass converting enzymes as industrial biocatalysts for fuels and chemicals: Recent developments. Catalysts 2012, 2, 244–263. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | Metric |
|---|---|
| Total reads | 345,471,086 |
| Total base (bp) | 51,820,662,900 |
| Number of contigs | 2,611,883 |
| Contig N50 (bp) | 1117 |
| Average contig length (bp) | 898 |
| Maximum contig length (bp) | 611,845 |
| Gene number | 4,104,872 |
| Gene N50 (bp) | 615 |
| Average gene length (bp) | 505 |
| Maximum gene length (bp) | 20,541 |
| Sources | Gene Num | Percentage (%) | Phylum | Class | Order | Family | Genus | Species |
|---|---|---|---|---|---|---|---|---|
| Bacteria | 3,884,879 | 99.69 | 111 | 83 | 170 | 406 | 1971 | 738 |
| Archaea | 293 | 0.01 | 9 | 12 | 18 | 23 | 50 | 8 |
| Eukaryota | 1144 | 0.03 | 7 | 26 | 46 | 79 | 113 | 86 |
| Viruses | 10,565 | 0.27 | 0 | 0 | 2 | 14 | 101 | 84 |
| Sum | 3,896,881 | 100 | 131 | 118 | 237 | 523 | 2240 | 916 |
| Total | Nr | Swissprot | KEGG | eggNOG | Overall | |
|---|---|---|---|---|---|---|
| ORFs | 4,104,872 | 3,923,046 | 2,382,630 | 2,809,791 | 3,279,853 | 3,925,740 |
| % | 100% | 95.57% | 58.04% | 68.45% | 79.90% | 95.64% |
| Cat * | Enzyme Name | ORF Number | Number of Complete ORFs Containing | ||
|---|---|---|---|---|---|
| (EC…) | Total | Com ** | Dom *** | Domain/Domain Types | |
| P | Pretreatment | 907 | 216 | 198 | 198/19 types |
| P1 | Pectinesterase (EC 3.1.1.11) | 815 | 199 | 181 | 61/CE8; 37/DUF4861; 29/CE2; 23/PL10; 15/Abhydrolase_3; 16/11 others |
| P4 | Feruloylesterase (EC 3.1.1.73) | 75 | 12 | 12 | 9/DUF3237; 3/Tannase |
| P3 | Laccase (EC 1.10.3.2) | 10 | 5 | 5 | 5/Cu3-Cu0-Cu2 |
| P4 | Expansin | 7 | 0 | 0 | |
| C | Cellulase | 8301 | 1279 | 1058 | 1058/81 types |
| C1 | β-glucosidase (EC 3.2.1.21) | 4272 | 503 | 475 | 220/GH3-FN3; 93/GH1; 29/FN3; 29/GH3; 20/GH43; 11/GH3-Exop_C; 10/DUF4886; 10/CE3; 53/19 others |
| C2 | Endoglucanase (EC 3.2.1.4) | 2216 | 548 | 367 | 105/GH8; 72/GH5; 38/PeptidaseM42; 18 GH5-CBM6; 14/DUF285; 13/GH18; 10/CE2; 97/43 others |
| C3 | 6-phospho-beta-glucosidase (EC 3.2.1.86) | 1718 | 213 | 210 | 152/GH1; 58/GH4 |
| C4 | Cellobiohydrolase (EC 3.2.1.91) | 73 | 15 | 6 | 1/Alginate_lyase; 1/Amidase 3; 1/CBM2; 1/CBP_BcsO; 1/GH128 + Laminin G3; 1/Znribbon8 |
| C5 | Cellobiose phosphorylase (EC 2.4.1.20) | 22 | 0 | 0 | |
| H | Hemicellulase | 13,018 | 2087 | 1828 | 1828/151 types |
| H1 | xyloglucan-active β-D-galactosidase (EC 3.2.1.23) | 3288 | 330 | 298 | 123/GH2; 28/GH42; 21/GH35; 20/DUF302; 18/GH43; 14/GH2 + CBM57; 13/Metallophos; 61/29 others |
| H2 | α-L-fucosidase (EC 3.2.1.51) | 2279 | 464 | 413 | 109/GH29; 81/GH95; 63/CE3; 46/Exo_endo_phos; 19/GH29 + CBM32; 16/CBM32; 13/GH33; 12/Big_2; 10/Abhydrolase_3; 9/GH117; 6/DUF1735 + CBM32; 29/19 others |
| H3 | α-galactosidase (EC 3.2.1.22) | 1033 | 163 | 134 | 65/GH36; 32/GH27; 16/GH4; 5/GH36 + GH27; 3/CBM51; 3/GH27 + CBM35; 2/Alginate_lyase; 8/8 others |
| H4 | α-L-arabinofuranosidase (EC 3.2.1.55) | 1016 | 169 | 161 | 59/CE3; 47/GH51; 46/GH43; 4/GH43 + CBM32; 3/GH43 + GH121; 1/GH54; 1/Methyltransf-23 |
| H5 | endo-β-1,4 xylanase (EC 3.2.1.8) | 885 | 230 | 175 | 65/Abhydrolase_3; 36/Peptidase_S9; 33/GH10; 15/CE15; 9/CBM6; 4/CE4; 13/9 others |
| H6 | alpha-D-xylosidexylohydrolase (EC 3.2.1.177) | 762 | 62 | 55 | 33/GH31; 9/GH31 + DUF5110; 6/Gal_mutarotas_2 + GH31; 2/DUF4968 + GH31 + DUF5110; 5/5 others |
| H7 | xylan 1,4-beta-xylosidase (EC 3.2.1.37) | 659 | 146 | 134 | 69/HTH_18; 45/GH43; 18/GH39; 2/AraC_binding + HTH_18 |
| H8 | Beta-mannosidase (EC 3.2.1.25) | 611 | 46 | 37 | 22/GH2; 10/GH2 + Ig; 4/Ig; 1/GH158 |
| H9 | oligosaccharide reducing-end xylanase (EC 3.2.1.156) | 552 | 100 | 73 | 31/GH43; 23/CHU; 4/GH8; 4/SprB; 3/CE4; 2/CBM9; 6/6 others |
| H10 | β-mannanase (EC 3.2.1.78) | 368 | 87 | 81 | 31/GH26; 17/GH5; 8/DUF1996; 7/GH44; 3/GH35; 3/CHU; 3/GH5 + CBM35; 9/9 others |
| H11 | Endopolygalacturonaselyase (EC 4.2.2.2) | 341 | 60 | 52 | 37/PL1; 3/PL1 + CBM77; 3/PL10; 3/PL2; 3/PL3; 2/CBM35 + PL1; 1/PL1 + LamininG3 |
| H12 | beta-fructofuranosidase (EC 3.2.1.26) | 255 | 38 | 36 | 31/GH32; 2/Big_2; 1/CBM38 + GH32; 1/GH137; 1/PAN_4 |
| H13 | beta-D-glucuronidase (EC 3.2.1.31) | 227 | 33 | 28 | 20/GH2; 6/GH141; 2/GH158 |
| H14 | Exopolygalacturonase (EC 3.2.1.67) | 223 | 74 | 69 | 67/GH28; 2/NAD_binding_10 |
| H15 | Licheninase (EC 3.2.1.73) | 175 | 52 | 52 | 48/GH16; 2/GH158 + GH16; 1/GH16 + CBM16; 1/GH16 + CBM6 |
| H16 | alpha-glucuronidase (EC 3.2.1.139) | 161 | 17 | 16 | 16/GH67 |
| H17 | Exopolygalacturonaselyase (EC 4.2.2.9) | 142 | 9 | 9 | 9/PL9 |
| H18 | Endopolygalacturonase (EC 3.2.1.15) | 38 | 6 | 4 | 4/GH28 |
| H19 | endo-transglycosylase/hydrolase (EC 2.4.1.207) | 2 | 1 | 1 | 1/GH16 |
| H20 | Acetylxylanesterase (EC 3.1.1.72) | 1 | 0 | 0 | |
| No | Enzyme Name (EC…) | Total | Complete | % Complete | Domain |
|---|---|---|---|---|---|
| 1 | Catalase/Peroxidase (EC 1.11.1.21) | 224 | 142 | 63% | Catalase |
| 2 | Feruloylesterase (EC 3.1.1.73) | 53 | 35 | 66% | Tannase |
| 3 | Multi-copper oxidase | 901 | 483 | 54% | Cu-oxidase, Cu_oxidase_2, Cu_oxidase_3, Cu_oxidase_4 |
| 4 | Lytic polysaccharide monooxygenase (EC 1.14.99.54) | 69 | 33 | 48% | LPMO_10 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Le, T.-T.-H.; Nguyen, T.-B.; Nguyen, H.-D.; Nguyen, H.-D.; Le, N.-G.; Dao, T.-K.; Nguyen, T.-Q.; Do, T.-H.; Truong, N.-H. De Novo Metagenomic Analysis of Microbial Community Contributing in Lignocellulose Degradation in Humus Samples Harvested from Cuc Phuong Tropical Forest in Vietnam. Diversity 2022, 14, 220. https://doi.org/10.3390/d14030220
Le T-T-H, Nguyen T-B, Nguyen H-D, Nguyen H-D, Le N-G, Dao T-K, Nguyen T-Q, Do T-H, Truong N-H. De Novo Metagenomic Analysis of Microbial Community Contributing in Lignocellulose Degradation in Humus Samples Harvested from Cuc Phuong Tropical Forest in Vietnam. Diversity. 2022; 14(3):220. https://doi.org/10.3390/d14030220
Chicago/Turabian StyleLe, Thi-Thu-Hong, Thi-Binh Nguyen, Hong-Duong Nguyen, Hai-Dang Nguyen, Ngoc-Giang Le, Trong-Khoa Dao, Thi-Quy Nguyen, Thi-Huyen Do, and Nam-Hai Truong. 2022. "De Novo Metagenomic Analysis of Microbial Community Contributing in Lignocellulose Degradation in Humus Samples Harvested from Cuc Phuong Tropical Forest in Vietnam" Diversity 14, no. 3: 220. https://doi.org/10.3390/d14030220
APA StyleLe, T.-T.-H., Nguyen, T.-B., Nguyen, H.-D., Nguyen, H.-D., Le, N.-G., Dao, T.-K., Nguyen, T.-Q., Do, T.-H., & Truong, N.-H. (2022). De Novo Metagenomic Analysis of Microbial Community Contributing in Lignocellulose Degradation in Humus Samples Harvested from Cuc Phuong Tropical Forest in Vietnam. Diversity, 14(3), 220. https://doi.org/10.3390/d14030220