
Ectomycorrhizospheric Microbiome Assembly Rules of Quercus mongolica in the Habitat of SongRong (Tricholoma matsutake) and the Effect of Neighboring Plants
Abstract
1. Introduction
2. Materials and Methods
2.1. Sampling Sites
2.2. Sample Collection and Processing
2.3. DNA Extraction
2.4. Determination of Rhizosphere Environment and Climatic Factors
2.5. Data Analyses
3. Results
3.1. Microbial Community from the Ectomycorrhizal Root Tips of Quercus mongolica
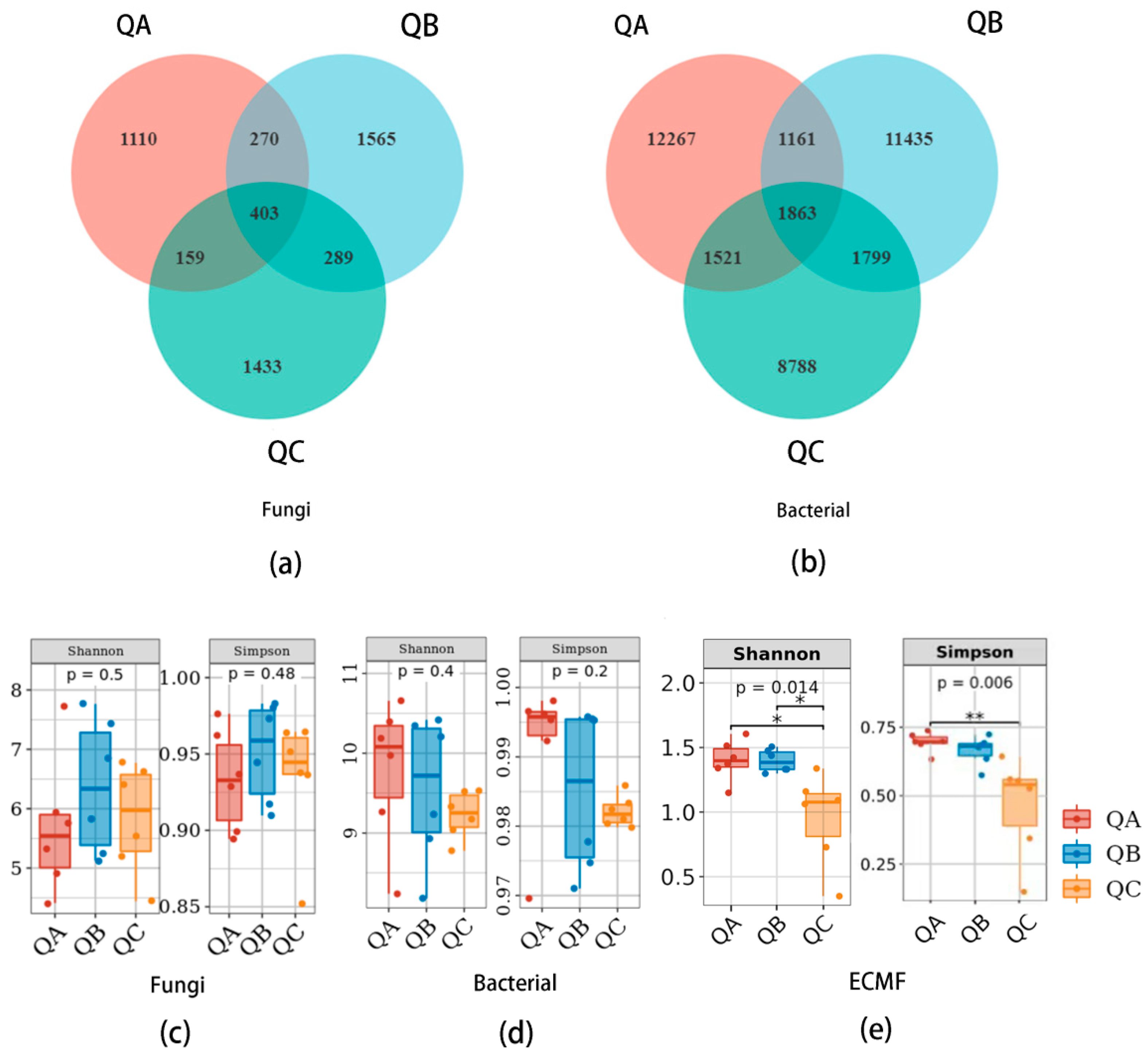
3.1.1. Alpha-Diversity of Fungal and Bacterial Communities
3.1.2. Beta-Diversity of Microbial Communities
3.1.3. Species Construction of Microbial Communities
3.2. Effects of Environmental Factors on Microbial Community in Ectomycorrhiza of Quercus mongolica
3.3. The Relationship between Microorganisms from the Ectomycorrhizal Root Tips of Quercus mongolica
3.4. The Predicted Metabolic Functions of Microorganisms from the Ectomycorrhizal Root Tips of Q. mongolica
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tedersoo, L.; Bahram, M.; Zobel, M. How mycorrhizal associations drive plant population and community biology. Science 2020, 367, eaba1223. [Google Scholar] [CrossRef] [PubMed]
- Rosenstock, N.P.; Berner, C.; Smits, M.M.; Krám, P.; Wallander, H. The role of phosphorus, magnesium and potassium availability in soil fungal exploration of mineral nutrient sources in Norway spruce forests. New Phytol. 2016, 211, 542–553. [Google Scholar] [CrossRef]
- Voříšková, J.; Brabcová, V.; Cajthaml, T.; Baldrian, P. Seasonal dynamics of fungal communities in a temperate oak forest soil. New Phytol. 2014, 201, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Ji, R.Q.; Zhou, J.J.; Ma, S.Y.; Xu, Y.; Li, Y. Investigation on Symbiotic Fungi in Roots of Coniferous Forests in Jingyuetan National Forest Park. J. Fungal Res. 2020, 18, 154–161. [Google Scholar]
- Smith, S.E.; Smith, F.A. Tansley review No. 20. structure and function of the interfaces in biotrophic symbioses as they relate to nutrient transport. New Phytol. 1990, 114, 1–38. [Google Scholar] [CrossRef] [PubMed]
- Trappe, J.M. A.B. Frank and mycorrhizae: The challenge to evolutionary and ecologic theory. Mycorrhiza 2005, 15, 277–281. [Google Scholar] [CrossRef]
- Elena, A.P.; Mikhail, N.G.; Svetlana, N.B.; Vera, F.M.; Ekaterina, F.M.; Alexander, E.K. Post-fire Successions of vegetation and Pinus koraiensis ectomycorrhizal communities in Korean pine–broadleaf forests of the central aikhote-alin. Achiev. Life Sci. 2016, 10, 48–56. [Google Scholar] [CrossRef]
- Southam, H.; Stafl, N.; Guichon, S.H.A.; Simard, S.W. Characterizing the Ectomycorrhizal Fungal Community of Whitebark Pine in Interior British Columbia: Mature Trees, Natural Regeneration and Planted Seedlings. Frontiers 2022, 4, 750701. [Google Scholar] [CrossRef]
- Ji, R.Q.; Gao, T.T.; Li, G.L.; Xu, Y.; Xing, P.J.; Zhou, J.J.; Xie, M.L.; Li, J.Q.; Li, Y. Correlation between ectomycorrhizal fungal community and environmental factors in Pinus koraiensis forest in Northeast China. Mycosystema 2020, 39, 743–754. [Google Scholar] [CrossRef]
- Dickie, I.A.; John, M.G.S. Molecular Mycorrhizal Symbiosis: Second-Generation Molecular Understanding of Mycorrhizas in Soil Ecosystems; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2017; pp. 474–491. [Google Scholar]
- Smith, S.E.; Read, D.J. Mycorrhizal Symbiosis; Academic Press: New York, NY, USA, 2008. [Google Scholar]
- Milović, M.; Orlović, S.; Grebenc, T.; Bajc, M.; Kovačević, B.; Kraigher, H. Ectomycorrhizal fungal community in mature white poplar plantation. Iforest-Biogeosciences For. 2021, 14, 540. [Google Scholar] [CrossRef]
- Amend, A.; Keeley, S.; Garbelotto, M. Forest age correlates with fine-scale spatial structure of Matsutake mycorrhizas. Mycol. Res. 2009, 113, 541–551. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, N.; Kikuchi, K.; Sasaki, Y.; Guerin-Laguette, A.; Vaario, L.-M.; Suzuki, K.; Lapeyrie, F.; Intini, M. Genetic relationship of Tricholoma matsutake and T. nauseosum from the Northern Hemisphere based on analyses of ribosomal DNA spacer regions. Mycoscience 2005, 46, 90–96. [Google Scholar] [CrossRef]
- Ogawa, M. Microbial ecology of mycorrhizal fungus Tricholoma matsutake (Ito et Imai) Sing. in pine forest. II. Mycorrhiza formed by Tricholoma matsutake. Bull. Gov. For. Exp. Sta. 1975, 272, 79–121. [Google Scholar]
- Li, Q.; Li, X.; Chen, C.; Li, S.; Huang, W.; Xiong, C.; Jin, X.; Zheng, L. Analysis of bacterial diversity and communities associated with Tricholoma matsutake fruiting bodies by barcoded pyrosequencing in Sichuan Province, Southwest China. Microbiol. Biotechnol. 2016, 26, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Park, M.S.; Oh, S.-Y.; Cho, H.J.; Fong, J.J.; Cheon, W.-J.; Lim, Y.W. Trichoderma songyi sp. nov., a new species associated with the pine mushroom (Tricholoma matsutake). Antonie Van Leeuwenhoek 2014, 106, 593–603. [Google Scholar] [CrossRef]
- Li, Q.; Li, X.; Huang, W.; Xiong, C.; Yang, Y.; Yang, Z.; Zheng, L. Community structure and diversity of entophytic bacteria in Tricholoma matsutake in Sichuan Province, Southwest China. Chin. J. Appl. Ecol. 2014, 25, 3316–3322. [Google Scholar]
- Liu, P.G.; Yuan, M.S.; Wang, X.H. Notes on the resources of matsutake group and their reasonable utilization as well as effective conservation in China. Jouranal Nat. Resour. 1999, 14, 245–252. [Google Scholar] [CrossRef]
- Fu, W.J.; Xu, G.B.; Liu, J.S. Research on Distribution and Ecological Environment of Tricholoma matsutake in Changbai Mountain Area. Acta Edulis Fungi 1998, 03, 48–52. [Google Scholar]
- Yang, M.H.; Yang, X.M.; Chen, L.G. Studies on the Relationship Between Tricholoma matsutake and Other Rhizosphere Microorganisms. Acta Agric. Univ. Jiangxiensis 1997, 4, 78–82. [Google Scholar]
- Ohara, H.; Hamada, M. Disappearance of bacteria from the zone of active mycorrhizas in Tricholoma matsutake (S. Ito et Imai) Singer. Nature 1967, 213, 528. [Google Scholar] [CrossRef]
- Yamanaka, T.; Yamada, A.; Furukawa, H. Advances in the cultivation of the highly-prized ectomycorrhizal mushroom Tricholoma matsutake. Mycoscience 2020, 61, 49–57. [Google Scholar] [CrossRef]
- Vaario, L.M.; Guerin-Laguette, A.; Gill, W.M.; Lapeyrie, F.; Suzuki, K. Only two weeks are required for Tricholoma matsutake to differentiate ectomycorrhizal Hartig net structures in roots of Pinus densiflora seedlings cultivated on artificial substrate. J. For. Res. 2000, 5, 293–297. [Google Scholar] [CrossRef]
- Guerin-Laguette, A.; Vaario, L.M.; Gill, W.M.; Lapeyrie, F.; Matsushita, N.; Suzuki, K. Rapid in vitro ectomycorrhizal infection on Pinus densiflora roots by Tricholoma matsutake. Mycoscience 2000, 41, 389–393. [Google Scholar] [CrossRef]
- Ka, K.-H.; Kim, H.-S.; Hur, T.-C.; Park, H.; Jeon, S.-M.; Ryoo, R.; Yeongseon, J. Analysis of environment and production of Tricholoma matsutake in Matsutake-infected pine trees (in Korean). Korean J. Mycol. 2018, 46, 34–42. [Google Scholar] [CrossRef]
- Yamada, A.; Endo, N.; Murata, H.; Ohta, A.; Fukuda, M. Tricholoma matsutake Y1 strain associated with Pinus densiflora shows a gradient of in vitro ectomycorrhizal specificity with Pinaceae and oak hosts. Mycoscience 2014, 55, 27–34. [Google Scholar] [CrossRef]
- Murata, H.; Babasaki, K.; Yamada, A. Highly polymorphic DNA markers to specify strains of the ectomycorrhizal basidiomycete Tricholoma matsutake based on σmarY1, the long terminal repeat of gypsy-type retroelement marY1. Mycorrhiza 2005, 15, 179–186. [Google Scholar] [CrossRef]
- Vaario, L.-M.; Pennanen, T.; Lu, J.; Palmén, J.; Stenman, J.; Leveinen, J.; Kilpeläinen, P.; Kitunen, V. Tricholoma matsutake can absorb and accumulate trace elements directly from rock fragments in the shiro. Mycorrhiza 2015, 25, 325–334. [Google Scholar] [CrossRef]
- Murata, H.; Yamada, A.; Maruyama, T.; Neda, H. Ectomycorrhizas in vitro between Tricholoma matsutake, a basidiomycete that associates with pinaceae, and Betula platyphylla var. japonica, an early-successional birch species, in cool-temperate forests. Mycorrhiza 2015, 25, 237–241. [Google Scholar] [CrossRef]
- Lian, C.; Hogetsu, T.; Matsushita, N.; Guerin-Laguette, A.; Suzuki, K.; Yamada, A. Development of microsatellite markers from an ectomycorrhizal fungus, Tricholoma matsutake, by an issr-suppression-pcr method. Mycorrhiza 2003, 13, 27–31. [Google Scholar] [CrossRef]
- Tan, W. Cultivation Theory and Method of Trichotoma matsutake. Acta Edulis Fungi 1994, 1, 53–63. [Google Scholar]
- Narimatsu, M.; Koiwa, T.; Masaki, T.; Sakamoto, Y.; Ohmori, H.; Tawaraya, K. Relationship between climate, expansion rate, and fruiting in fairy rings (‘shiro’) of an ectomycorrhizal fungus Tricholoma matsutake in a Pinus densiflora forest. Fungal Ecol. 2015, 15, 18–28. [Google Scholar] [CrossRef]
- Li, W.G.; Zhao, Y.G. Ecological conditions suitable for growth of Trichotoma matsutake. J. Microbiol. 2000, 20, 52–54. [Google Scholar]
- Xu, G.B.; Fu, W.J.; Zhang, M.J. Ecological Behaviour and Domestication of Tricholoma Matsutake in Changbai Mountain Area. J. Agric. Sci. Yanbian Univ. 1999, 2, 130–135. [Google Scholar]
- Xing, P.J.; Xu, Y.; Gao, T.T.; Li, G.L.; Zhou, J.J.; Xie, M.L.; Ji, R.Q. The community composition variation of Russulaceae associated with the Quercus mongolica forest during the growing season at Wudalianchi City, China. PeerJ 2020, 8, e8527. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Yu, K.; Chen, B.; Liao, Z.H.; Liang, J.Y.; Yao, Q.C.; Qin, Z.J.; Wang, H.; Yu, J. Different responses of scleractinian coral Acropora pruinosa from Weizhou Island during extreme high temperature events. Coral Reefs 2021, 40, 1697–1711. [Google Scholar] [CrossRef]
- White, T.J.; Bruns, T.; Lee, S.; Taylor, J. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In PCR Protocols: A Guide to Methods and Applications; Innis, M.A., Gelfand, D.H., Sninsky, J.J., White, T.J., Eds.; Academic Press: San Diego, CA, USA, 1990; pp. 315–322. [Google Scholar]
- Bao, S.D. Soil Agrochemical Analysis, 3rd ed.; China Agricultural Press: Beijing, China, 2000; pp. 1–495. [Google Scholar]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.; Al-Ghalith, G.A.; Caporaso, J.G. QIIME 2: Reproducible, interactive, scalable, and extensible microbiome data science. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2015, 13, 581–583. [Google Scholar] [CrossRef]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree: Computing large minimum evolution trees with profiles instead of a distance matrix. Mol. Biol. Evol. 2009, 26, 1641–1650. [Google Scholar] [CrossRef]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef]
- Nicholas, B.; Benjamin, D.K.; Ram, R.J.; Matthew, D.; Evan, B.; Rob, K.; Gavin, A.H.; Gregory, C.J. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2′s q2-feature-classifier plugin. Microbiome 2018, 6, 90. [Google Scholar] [CrossRef]
- Kõljalg, U.; Nilsson, R.H.; Abarenkov, K.; Tedersoo, L.; Taylor, A.F.S.; Bahram, M.; Bates, S.T.; Bruns, T.D.; Bengtsson-Palme, J.; Callaghan, T.M. Towards a unified paradigm for sequence-based identification of fungi. Mol. Ecol. 2013, 22, 5271–5277. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing [Computer Software Manual]; R Core Team: Vienna, Austria, 2019. [Google Scholar]
- Whittaker, R.H. Vegetation of the Siskiyou Mountains, Oregon and California. Ecol. Monogr. 1960, 30, 279–338. [Google Scholar] [CrossRef]
- Whittaker, R.H. Evolution and measurement of species diversity. Taxon 1972, 21, 213–251. [Google Scholar] [CrossRef]
- Shannon, C.E. A mathematical theory of communication. Bell Syst. Tech. J. 1948, 27, 379–423. [Google Scholar] [CrossRef]
- Simpson, E.H. Measurement of diversity. Nature 1949, 163, 688. [Google Scholar] [CrossRef]
- Yang, Y.; Yan, W.; Wei, J. Ectomycorrhizal fungal community in the rhizospheric soil of Betula platyphylla in Inner Mongolia. Mycosystema 2018, 37, 294–304. [Google Scholar]
- Clarke, K.R. Non-parametric multivariate analyses of changes in community structure. Aust. J. Ecol. 1993, 18, 117–143. [Google Scholar] [CrossRef]
- Faust, K.; Raes, J. Microbial interactions: From networks to models. Nat. Rev. Microbiol. 2012, 10, 538–550. [Google Scholar] [CrossRef]
- Douglas, G.M.; Maffei, V.; Zaneveld, J.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.I. PICRUSt2: An improved and extensible approach for metagenome inference. bioRxiv 2019, 10, 672295. [Google Scholar]
- Suz, L.M.; Kallow, S.; Reed, K.; Bidartondo, M.I.; Barsoum, N. Pine mycorrhizal communities in pure and mixed pine-oak forests: Abiotic environment trumps neighboring oak host effects. For. Ecol. Manag. 2017, 406, 370–380. [Google Scholar] [CrossRef]
- Xiong, C.; Zhu, Y.G.; Wang, J.T.; Singh, B.; Han, L.L.; Shen, J.P.; Li, P.P.; Wang, G.B.; Wu, C.F.; Ge, A.H.; et al. Host selection shapes crop microbiome assembly and network complexity. New Phytol. 2021, 229, 1091–1104. [Google Scholar] [CrossRef] [PubMed]
- Mony, C.; Gaudu, V.; Ricono, C.; Jambon, O.; Vandenkoornhuyse, P. Plant neighbours shape fungal assemblages associated with plant roots: A new understanding of niche-partitioning in plant communities. Funct. Ecol. 2021, 35, 1768–1782. [Google Scholar] [CrossRef]
- Gao, C.; Montoya, L.; Xu, L.; Madera, M.; Hollingsworth, J.; Purdom, E.; Singan, V.; Vogel, J.; Hutmacher, R.B.; Dahlberg, J.A.; et al. Fungal community assembly in drought-stressed sorghum shows stochasticity, selection, and universal ecological dynamics. Nat. Commun. 2020, 11, 1–14. [Google Scholar] [CrossRef]
- Chaluvadi, S.; Bennetzen, J.L. Species-associated differences in the below-ground microbiomes of wild and domesticated Setaria. Front. Plant Sci. 2018, 9, 1183. [Google Scholar] [CrossRef]
- Wang, Q.F.; Ma, M.C.; Jiang, X.; Zhou, B.K.; Guan, D.W.; Cao, F.M.; Chen, S.F.; Li, J. Long-term N fertilization altered 13C-labeled fungal community composition but not diversity in wheat rhizosphere of Chinese black soil. Soil Biol. Biochem. 2019, 135, 117–126. [Google Scholar] [CrossRef]
- Tan, S.; Liu, J.; Fang, Y.; Hedlund, B.P.; Lian, Z.H.; Huang, L.Y.; Li, J.T.; Huang, L.N.; Li, W.J.; Jiang, H.C.; et al. Insights into ecological role of a new deltaproteobacterial order Candidatus Acidulodesulfobacterales by metagenomics and metatranscriptomics. ISME J. 2019, 13, 2044–2057. [Google Scholar] [CrossRef]
- Pang, Z.Q.; Xu, P.; Yu, D.Q. Environmental adaptation of the root microbiome in two rice ecotypes. Microbiol. Res. 2020, 241, 126588. [Google Scholar] [CrossRef]
- Zhou, W.H.; Wang, Y.T.; Lian, Z.H.; Yang, T.T.; Zeng, Q.W.; Feng, S.W.; Fang, Z.; Shu, W.S.; Huang, L.N.; Ye, Z.H.; et al. Revegetation approach and plant identity unequally affect structure, ecological network and function of soil microbial community in a highly acidified mine tailings pond. Sci. Total Environ. 2020, 744, 140793. [Google Scholar] [CrossRef]
- Badri, D.V.; De-la-Peña, C.; Lei, Z.; Manter, D.K.; Chaparro, J.M.; Guimarães, R.L.; Sumner, L.W.; Vivanco, J.M. Root secreted metabolites and proteins are involved in the early events of plant-plant recognition prior to competition. PLoS ONE 2012, 7, e46640. [Google Scholar] [CrossRef]
- Pierik, R.; Mommer, L.; Voesenek, L.A. Molecular mechanisms of plant competition: Neighbour detection and response strategies. Funct. Ecol. 2013, 27, 841–853. [Google Scholar] [CrossRef]
- Lumibao, C.Y.; Kimbrough, E.; Formel, S.; Day, R.R.; From, A.S.; Conner, W.H.; Krauss, K.W.; Bael, S.A.V. Salinity, water level, and forest structure contribute to baldcypress (Taxodium distichum) rhizosphere and endosphere community structure. Wetlands 2020, 40, 2179–2188. [Google Scholar] [CrossRef]
- Ulbrich, T.C.; Rivas-Ubach, A.; Tiemann, L.K.; Friesen, M.L.; Evans, S.E. Plant root exudates and rhizosphere bacterial communities shift with neighbor context. Soil Biol. Biochem. 2022, 172, 108753. [Google Scholar] [CrossRef]
- Shen, X.F.; Fang, Y.; Dong, Z.X. Effects of sugarcane/peanut intercropping on soil microbes and soil enzyme activities. Crops 2014, 5, 55–58. [Google Scholar]
- Walters, W.A.; Jin, Z.; Youngblut, N.; Wallace, J.G.; Sutter, J.; Zhang, W.; González-Peña, A.; Peiffer, J.; Koren, O.; Shi, Q.; et al. Large-scale replicated field study of maize rhizosphere identifies heritable microbes. Proc. Natl. Acad. Sci. USA 2018, 115, 7368–7373. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Naylor, D.; Dong, Z.B.; Simmons, T.; Pierroz, G.; Hixson, K.K.; Kim, Y.M.; Zink, E.M.; Engbrecht, K.M.; Wang, Y.; et al. Drought delays development of the sorghum root microbiome and enriches for monoderm bacteria. Proc. Natl. Acad. Sci. USA 2018, 115, E4284–E4293. [Google Scholar] [CrossRef]
- Ashhab, A.A.; Meshner, S.; Alexander-Shani, R.; Dimerets, H.; Brandwein, M.; Bar-Lavan, Y.; Winters, G. Temporal and Spatial Changes in Phyllosphere Microbiome of Acacia Trees Growing in Arid Environments. Front. Microbiol. 2021, 12, 656269. [Google Scholar] [CrossRef]
- Zhang, R.F.; Vivanco, J.M.; Shen, Q.R. The unseen rhizosphere root-soil-microbe interactions for crop production. Curr. Opin. Microbiol. 2017, 37, 8–14. [Google Scholar] [CrossRef]
- Heijden, G.A.; Bruin, S.D.; Luckerhoff, L.; Logtestijn, R.S.P.; Schlaeppi, K. A widespread plant-fungal-bacterial symbiosis promotes plant biodiversity, plant nutrition and seedling recruitment. ISME J. 2016, 10, 389–399. [Google Scholar] [CrossRef]
- Vandenkoornhuyse, P.; Quaiser, A.; Duhamel, M.; Van, A.L.; Dufresne, A. The importance of the microbiome of the plant holobiont. New Phytol. 2015, 206, 1196–1206. [Google Scholar] [CrossRef]
- Jacoby, R.; Peukert, M.; Succurro, A.; Koprivova, A.; Kopriva, S. The role of soil microorganisms in plant mineral nutrition—Current knowledge and future directions. Front. Plant Sci. 2017, 8, 1617. [Google Scholar] [CrossRef] [PubMed]
- Rolfe, S.A.; Griffiths, J.; Ton, J. Crying out for help with root exudates: Adaptive mechanisms by which stressed plants assemble health-promoting soil microbiomes. Curr. Opin. Microbiol. 2019, 49, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Frew, A.; Wilson, B.A.L. Different mycorrhizal fungal communities differentially affect plant phenolic-based resistance to insect herbivory. Rhizosphere 2021, 19, 100365. [Google Scholar] [CrossRef]
- Ji, R.Q.; Xu, Y.; Si, Y.J.; Phukhamsakda, C.; Li, Y.; Meng, L.P.; Liu, S.Y.; Xie, M.L. Fungal–Bacterial Networks in the Habitat of SongRong (Tricholoma matsutake) and Driving Factors of Their Distribution Rules. J. Fungi 2022, 8, 575. [Google Scholar] [CrossRef] [PubMed]
- Redford, A.J.; Bowers, R.M.; Knight, R.; Linhart, Y.; Fierer, N. The ecology of the phyllosphere: Geographic and phylogenetic variability in the distribution of bacteria on tree leaves. Environ. Microbiol. 2010, 12, 2885–2893. [Google Scholar] [CrossRef]
- Balint, M.; Bartha, L.; O′Hara, R.B.; Olson, M.S.; Otte, J.; Pfenninger, M.; Robertson, A.L.; Tiffin, P.; Schmitt, I. Relocation, highlatitude warming and host genetic identity shape the foliar fungal microbiome of poplars. Mol. Ecol. 2015, 24, 235–248. [Google Scholar] [CrossRef]
- Rostas, M.; Cripps, M.G.; Silcock, P. Aboveground endophyte affects root volatile emission and host plant selection of a belowground insect. Oecologia 2015, 177, 487–497. [Google Scholar] [CrossRef]
- Schmidt, J.E.; Kent, A.D.; Brisson, V.L.; Gaudin, A.C.M. Agricultural management and plant selection interactively affect rhizosphere microbial community structure and nitrogen cycling. Microbiome 2019, 7, 146. [Google Scholar] [CrossRef]
- Philippot, L.; Raaijmakers, J.M.; Lemanceau, P.; van der Putten, W.H. Going back to the roots: The microbial ecology of the rhizosphere. Nat. Rev. Microbiol. 2013, 11, 789–799. [Google Scholar] [CrossRef]
- Apigo, A.; Oono, R. Dimensions of host specificity in foliar fungal endophytes. In Endophytes of Forest Trees; Pirttil, A., Frank, A., Eds.; Springer: Dordrecht, The Netherlands, 2018; pp. 15–42. [Google Scholar] [CrossRef]
- U’Ren, J.M.; Lutzoni, F.; Miadlikowska, J.; Zimmerman, N.B.; Carbone, J.; May, G.; Arnold, A.E. Host availability drives distributions of fungal endophytes in the imperilled boreal realm. Nat. Ecol. Evol. 2019, 3, 1430–1437. [Google Scholar] [CrossRef]
- Xiong, W.; Jousset, A.; Guo, S.; Karlsson, I.; Zhao, Q.Y.; Wu, H.S.; Kowalchuk, G.A.; Shen, Q.R.; Li, R.; Geisen, S. Soil protist communities form a dynamic hub in the soil microbiome. ISME J. 2018, 12, 634–638. [Google Scholar] [CrossRef] [PubMed]
- Yurgel, S.N.; Douglas, G.M.; Dusault, A.; Percival, D.; Langille, M.G.I. Dissecting community structure in wild blueberry root and soil microbiome. Front. Microbiol. 2018, 9, 1187. [Google Scholar] [CrossRef] [PubMed]
- Schoener, T.W. Resource partitioning in ecological communities. Science 1974, 185, 27–39. [Google Scholar] [CrossRef]
- Wu, S.H.; Huang, B.H.; Huang, C.L.; Li, G.; Liao, P.C. The aboveground vegetation type and underground soil property mediate the divergence of soil microbiomes and the biological interactions. Microb. Ecol. 2018, 75, 434–446. [Google Scholar] [CrossRef]
- Wei, Y.; Liu, X.; Ge, S.; Zhang, H.; Che, X.; Liu, S.; Liu, D.; Li, H.; Gu, H.; He, L.; et al. Involvement of Phospholipase C in Photosynthesis and Growth of Maize Seedlings. Genes 2022, 13, 1011. [Google Scholar] [CrossRef] [PubMed]
- Wagner, G.; Hollmann, S. Uronic acid dehydrogenase from Pseudomonas syringae. Purif. Prop. 1976, 61, 589–596. [Google Scholar] [CrossRef]
- Harry, B.; Hannu, M.; Anu, K.; Merja, P.; Peter, R. Identification in Agrobacterium tumefaciens of the D-galacturonic acid dehydrogenase gene. Appl. Microbiol. Biotechnol. 2010, 86, 901–909. [Google Scholar]
- Johannes, G.; Annette, Z.; Magdy, M.; Hermann, S.; Georg, F. Genes coding for a new pathway of aerobic benzoate metabolism in Azoarcus evansii. J. Bacteriol. 2002, 184, 6301–6315. [Google Scholar] [CrossRef]
- Uwe, A.; Brigitte, O.; Erdmute, S.; Heidrun, H.; Georg, F. New aerobic benzoate oxidation pathway via benzoyl-coenzyme A and 3-hydroxybenzoyl-coenzyme A in a denitrifying Pseudomonas sp. J. Bacteriol. 1993, 175, 4851–4858. [Google Scholar] [CrossRef][Green Version]
- Jasleen, B.; Martin, J.B. Structural and biochemical characterization of a novel aldehyde dehydrogenase encoded by the benzoate oxidation pathway in Burkholderia xenovorans LB400. J. Mol. Biol. 2008, 379, 597–608. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fungi | Bacteria | Ectomycorrhizal Fungi | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Df | R2 | Pr (>F) | Df | R2 | Pr (>F) | Df | R2 | Pr (>F) | |
| QA/QB/QC | 2 | 0.3721 | 0.001 | 2 | 0.38946 | 0.001 | 2 | 0.49682 | 0.001 |
| QB/QA | 1 | 0.31335 | 0.002 | 1 | 0.28644 | 0.002 | 1 | 0.47184 | 0.006 |
| QC/QA | 1 | 0.26416 | 0.005 | 1 | 0.35696 | 0.002 | 1 | 0.37081 | 0.001 |
| QC/QB | 1 | 0.34905 | 0.004 | 1 | 0.33499 | 0.004 | 1 | 0.45186 | 0.004 |
| Sample Soil ID | SpH (mg·kg−1) | SOM (mg·kg−1) | SAK (mg·kg−1) | SEN (mg·kg−1) | SAP (mg·kg−1) | T (°C) | RM (mm) |
|---|---|---|---|---|---|---|---|
| Sample plot A | 5.71 ± 0.11a | 106.89 ± 23.26a | 23.57 ± 8.26a | 203.73 ± 22.68a | 20.19 ± 15.54a | 19.67 ± 2a | 10.05 ± 8.87a |
| Sample plot B | 5 ± 0.19c | 68.04 ± 12.54b | 25.83 ± 1.43a | 114.32 ± 10.98b | 4.67 ± 3.46b | 19.67 ± 2a | 10.05 ± 8.87a |
| Sample plot C | 5.29 ± 0.1b | 61.32 ± 6.15b | 20.27 ± 4.54a | 94.82 ± 7.49c | 3.43 ± 2.69b | 19.03 ± 0.86a | 1.63 ± 3.71a |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Si, Y.-J.; Xu, Y.; Li, B.-Q.; Liu, J.; Meng, L.-P.; Li, Y.; Ji, R.-Q.; Liu, S.-Y. Ectomycorrhizospheric Microbiome Assembly Rules of Quercus mongolica in the Habitat of SongRong (Tricholoma matsutake) and the Effect of Neighboring Plants. Diversity 2022, 14, 810. https://doi.org/10.3390/d14100810
Si Y-J, Xu Y, Li B-Q, Liu J, Meng L-P, Li Y, Ji R-Q, Liu S-Y. Ectomycorrhizospheric Microbiome Assembly Rules of Quercus mongolica in the Habitat of SongRong (Tricholoma matsutake) and the Effect of Neighboring Plants. Diversity. 2022; 14(10):810. https://doi.org/10.3390/d14100810
Chicago/Turabian StyleSi, Yan-Ji, Yang Xu, Bin-Qi Li, Jin Liu, Li-Peng Meng, Yu Li, Rui-Qing Ji, and Shu-Yan Liu. 2022. "Ectomycorrhizospheric Microbiome Assembly Rules of Quercus mongolica in the Habitat of SongRong (Tricholoma matsutake) and the Effect of Neighboring Plants" Diversity 14, no. 10: 810. https://doi.org/10.3390/d14100810
APA StyleSi, Y.-J., Xu, Y., Li, B.-Q., Liu, J., Meng, L.-P., Li, Y., Ji, R.-Q., & Liu, S.-Y. (2022). Ectomycorrhizospheric Microbiome Assembly Rules of Quercus mongolica in the Habitat of SongRong (Tricholoma matsutake) and the Effect of Neighboring Plants. Diversity, 14(10), 810. https://doi.org/10.3390/d14100810