
Bacterial Diversity and Community in Regional Water Microbiota between Different Towns in World’s Longevity Township Jiaoling, China


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. DNA Extraction and Polymerase Chain Reaction (PCR) Amplification
2.3. 16S rRNA Gene Sequencing
2.4. Processing of Sequencing Data
2.5. Statistical Analysis
3. Results
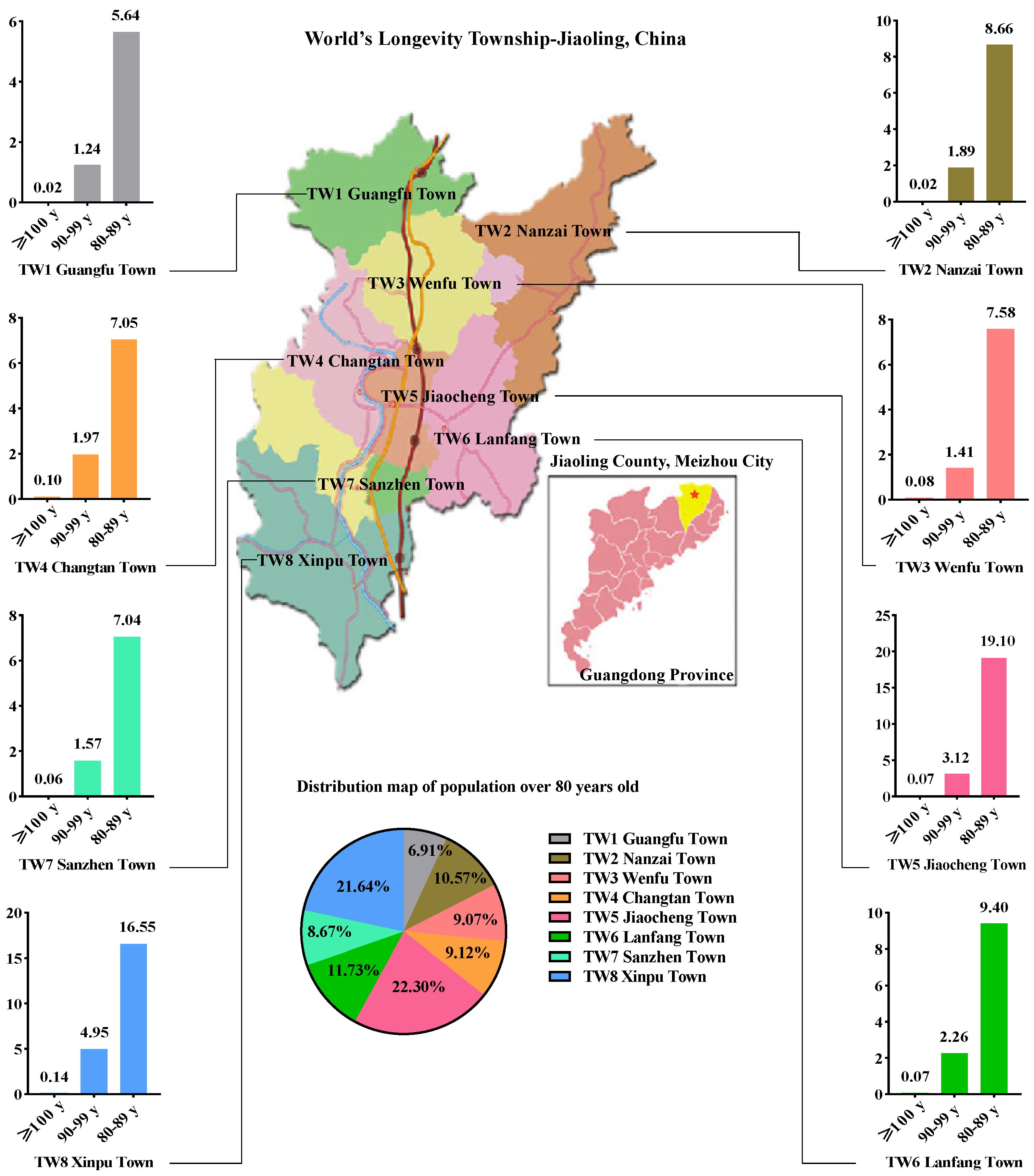
3.1. Characteristics of the Longevous Populations and Sample Sequencing
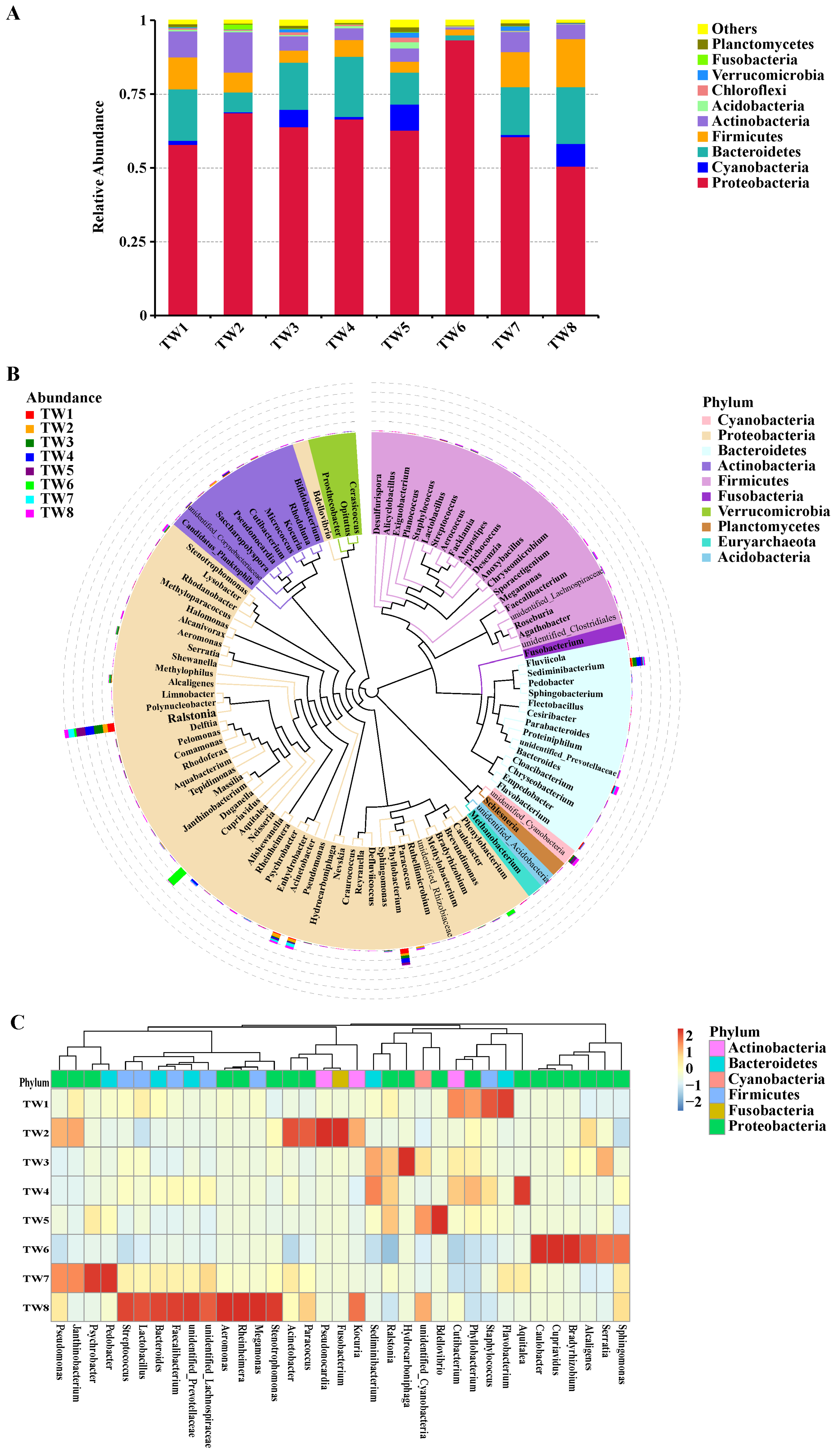
3.2. Composition and Structure of the Water Microbiota
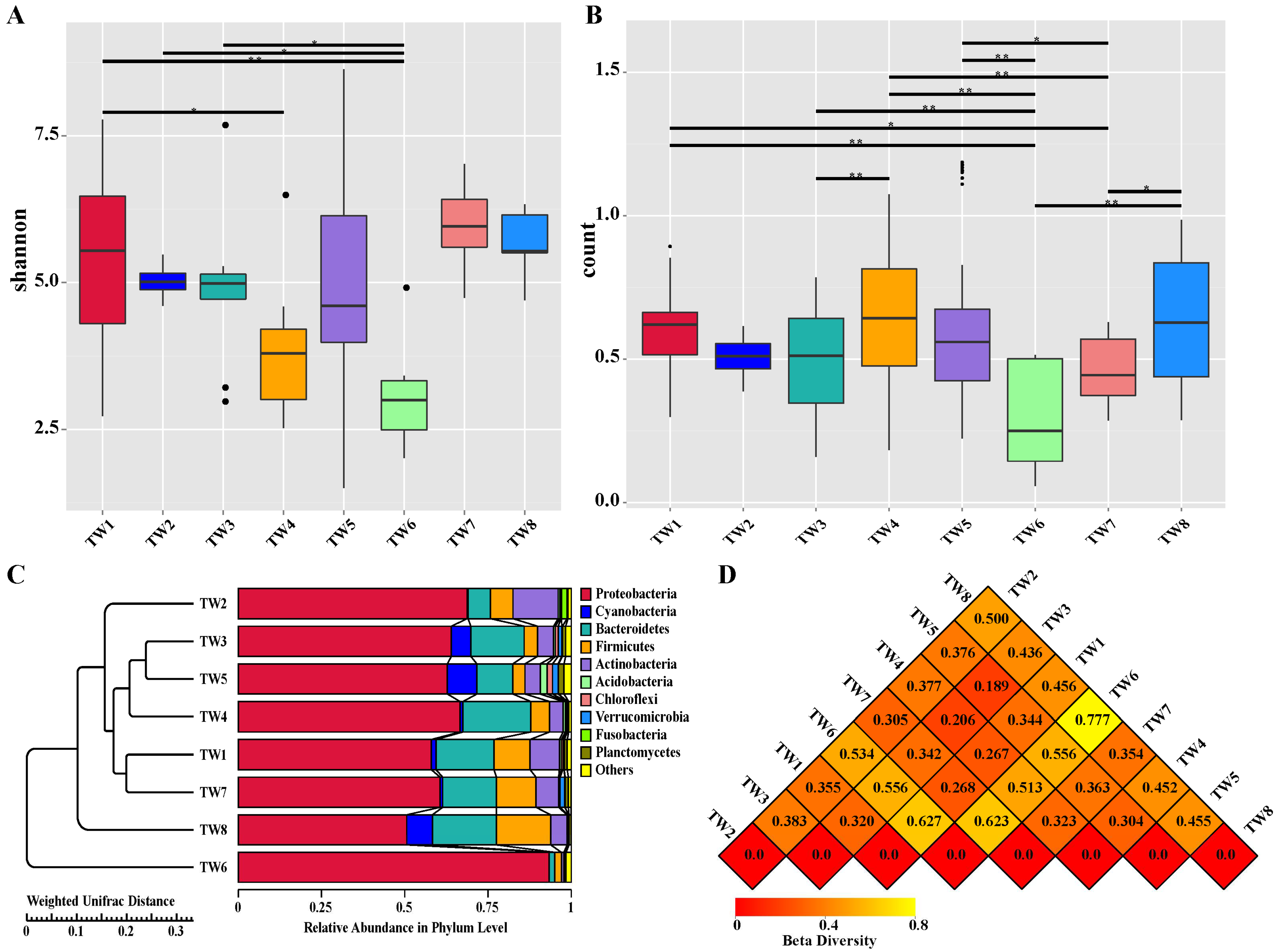
3.3. Alpha Diversity and Beta Diversity Analyses
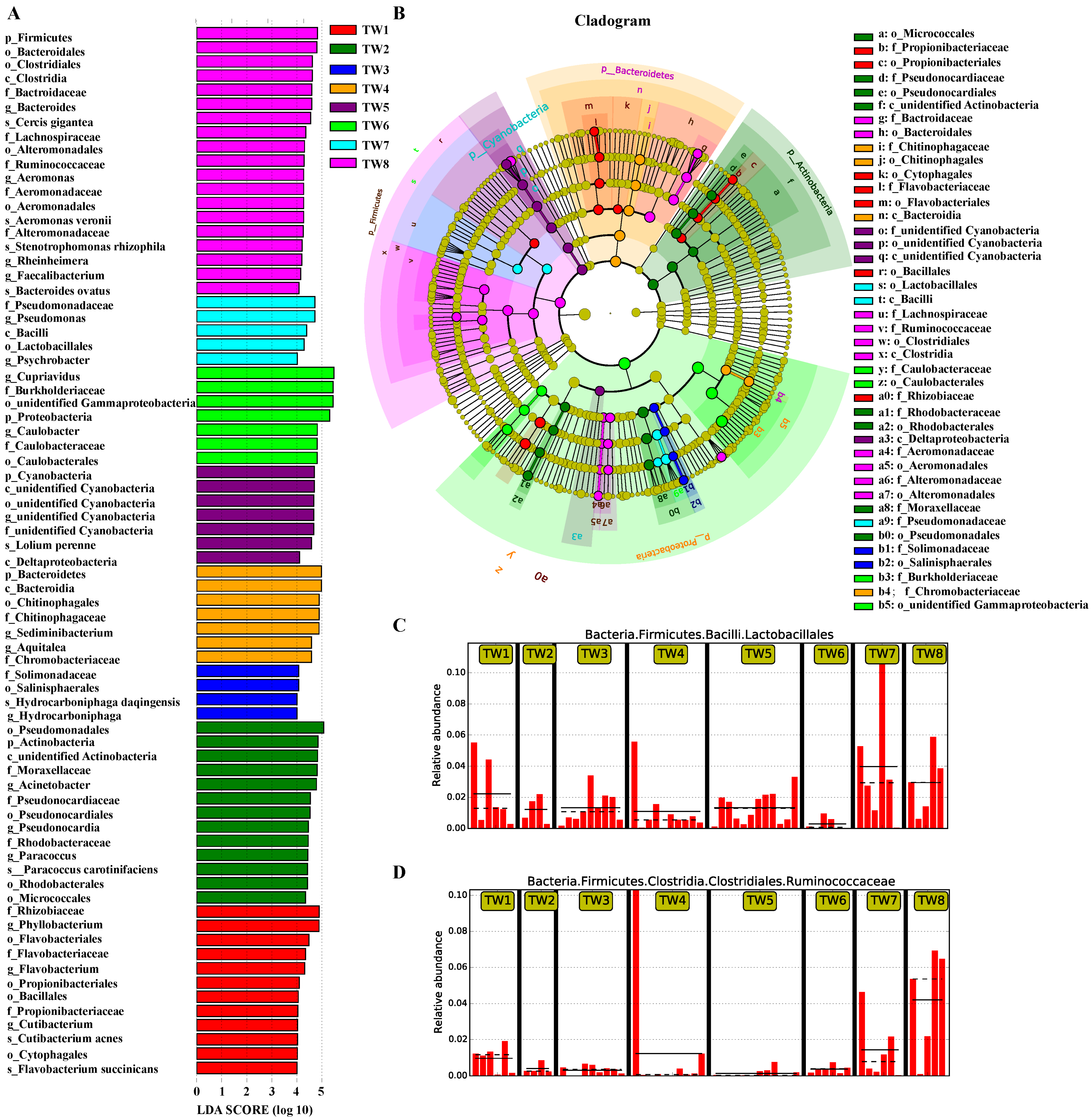
3.4. Significance Analysis of Species Differences between Groups
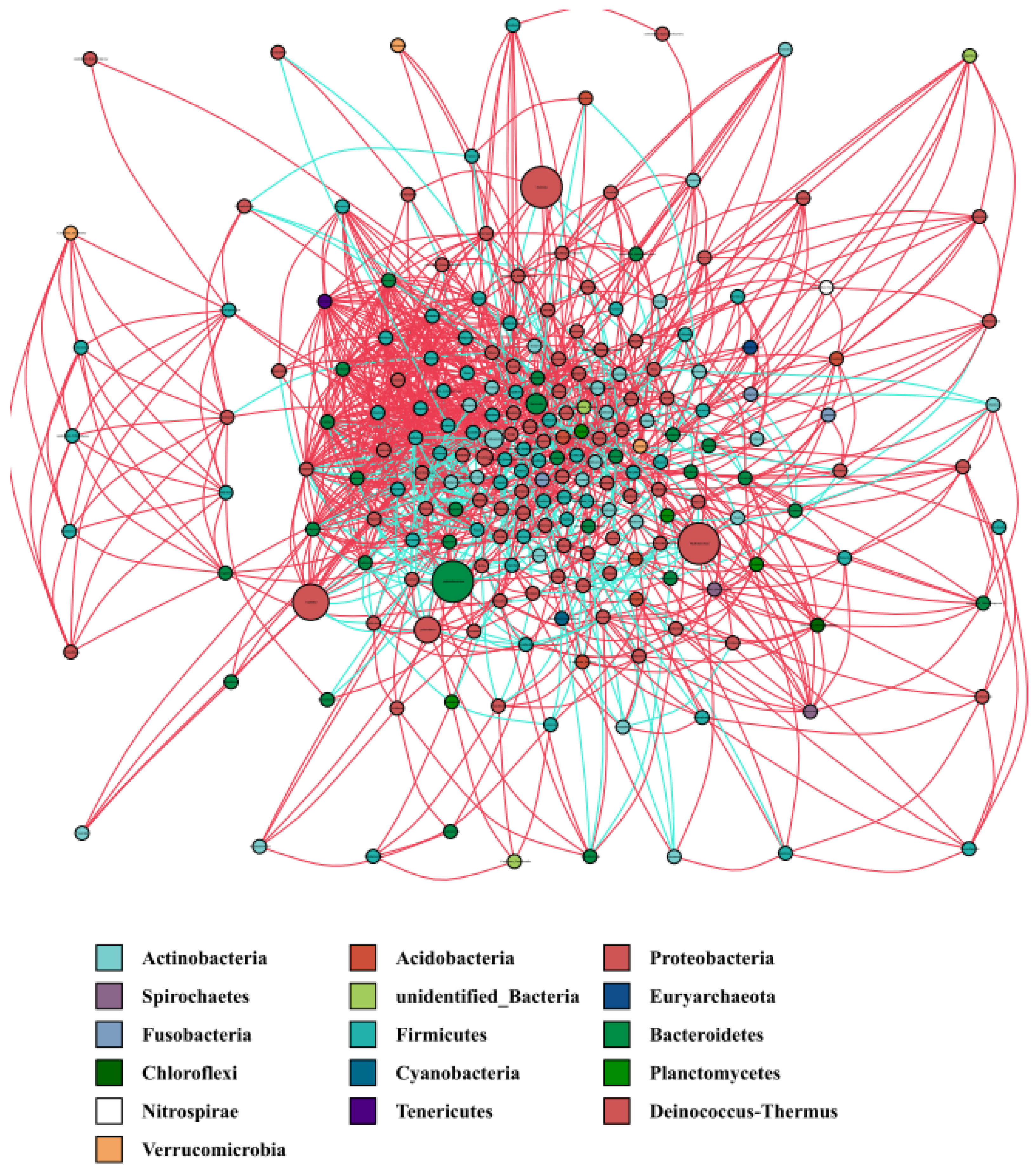
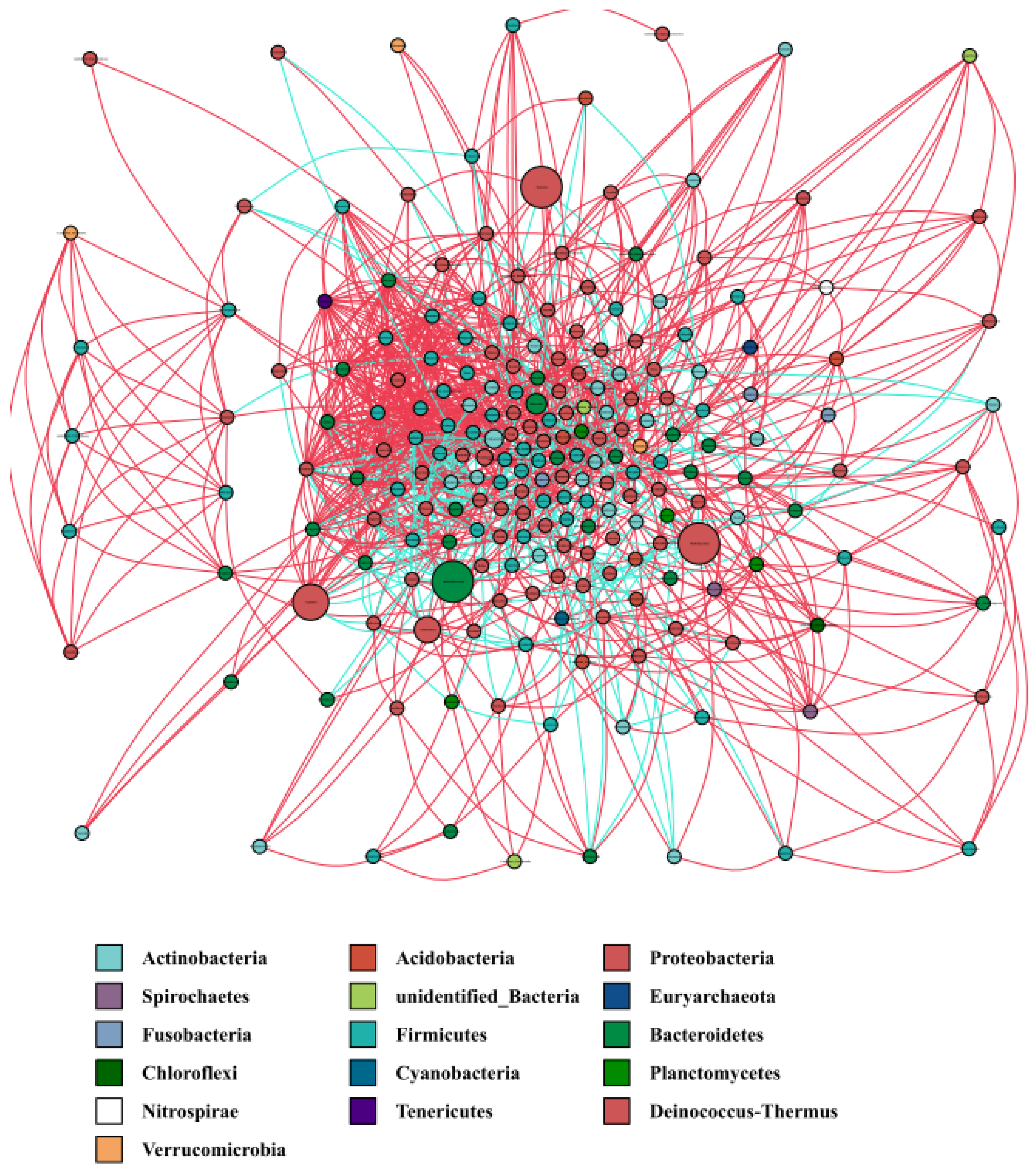
3.5. Co-Occurrence Network of the Water Microbiota
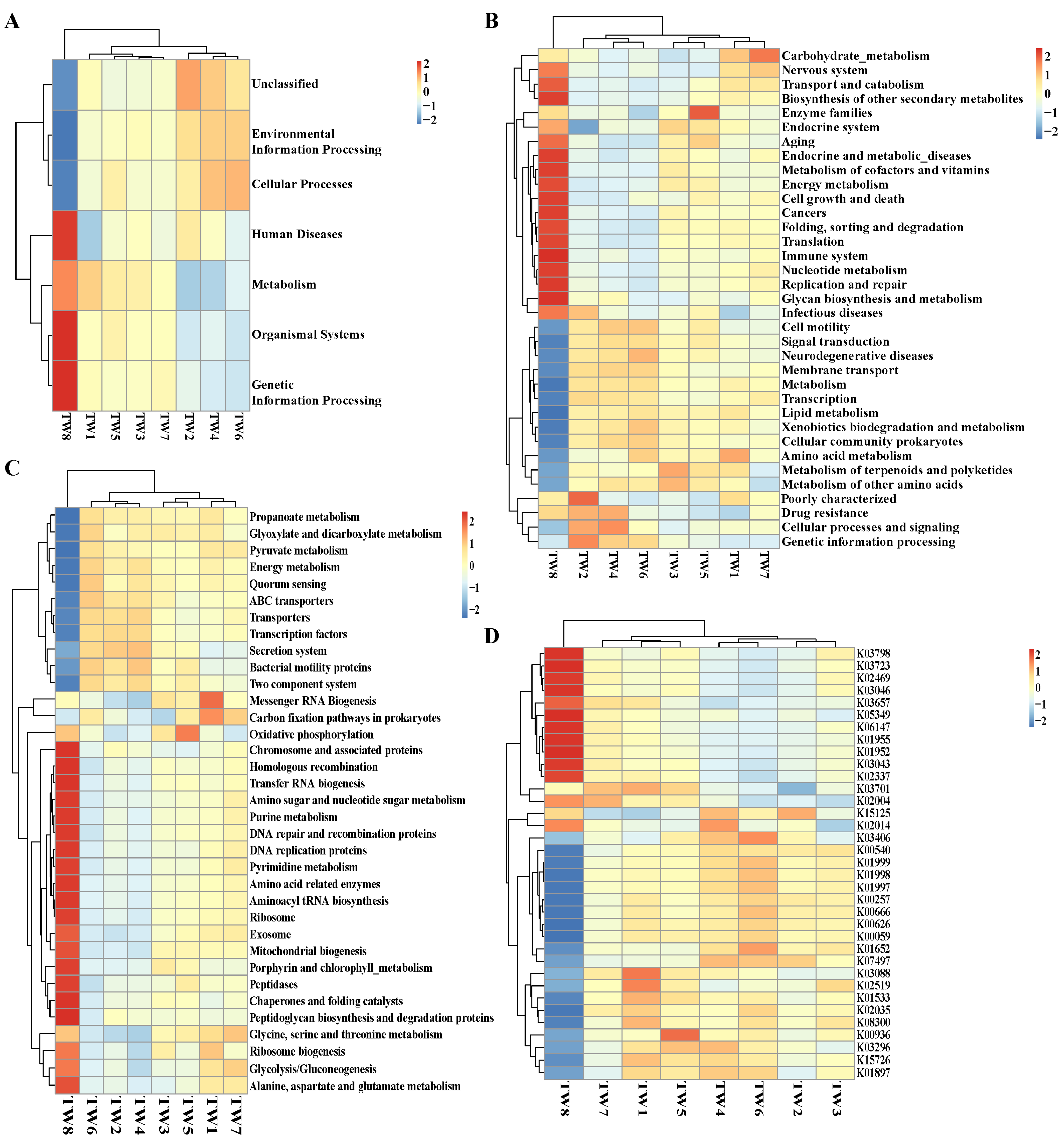
3.6. Predicted Functions of the Water Microbiota
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Barak, Y.; Leitch, S.; Glue, P. The Great Escape. Centenarians’ exceptional health. Aging Clin. Exp. Res. 2020, 33, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Balistreri, C.; Candore, G.; Accardi, G.; Buffa, S.; Bulati, M.; Martorana, A.; Colonna-Romano, G.; Lio, D.; Caruso, C. Centenarian offspring: A model for understanding longevity. Curr. Vasc. Pharmacol. 2013, 12, 718–725. [Google Scholar] [CrossRef] [PubMed]
- Costa, D.; Scognamiglio, M.; Fiorito, C.; Benincasa, G.; Napoli, C. Genetic background, epigenetic factors and dietary interventions which influence human longevity. Biogerontology 2019, 20, 605–626. [Google Scholar] [CrossRef] [PubMed]
- Gruber, J.; Kennedy, B.K. Microbiome and Longevity: Gut Microbes Send Signals to Host Mitochondria. Cell 2017, 169, 1168–1169. [Google Scholar] [CrossRef] [Green Version]
- Han, B.; Sivaramakrishnan, P.; Lin, C.-C.J.; Neve, I.A.; He, J.; Tay, L.W.R.; Sowa, J.N.; Sizovs, A.; Du, G.; Wang, J.; et al. Microbial Genetic Composition Tunes Host Longevity. Cell 2017, 169, 1249–1262.e13. [Google Scholar] [CrossRef] [Green Version]
- Caruso, C.; Aiello, A.; Accardi, G.; Ciaglia, E.; Cattaneo, M.; Puca, A. Genetic Signatures of Centenarians: Implications for Achieving Successful Aging. Curr. Pharm. Des. 2019, 25, 4133–4138. [Google Scholar] [CrossRef]
- Kenyon, C.J. The genetics of ageing. Nat. Cell Biol. 2010, 464, 504–512. [Google Scholar] [CrossRef]
- Melzer, D.; Pilling, L.C.; Ferrucci, L. The genetics of human ageing. Nat. Rev. Genet. 2019, 21, 88–101. [Google Scholar] [CrossRef]
- Joshi, P.K.; Pirastu, N.; Kentistou, K.A.; Fischer, K.; Hofer, E.; Schraut, K.E.; Clark, D.W.; Nutile, T.; Barnes, C.L.K.; Timmers, P.R.H.J.; et al. Genome-wide meta-analysis associates HLA-DQA1/DRB1 and LPA and lifestyle factors with human longevity. Nat. Commun. 2017, 8, 910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Partridge, L.; Deelen, J.; Slagboom, P.E. Facing up to the global challenges of ageing. Nat. Cell Biol. 2018, 561, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Bektas, A.; Schurman, S.; Sen, R.; Ferrucci, L. Aging, inflammation and the environment. Exp. Gerontol. 2018, 105, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Pan, C.; Fan, Q.; Yang, J.; Deng, D. Health Inequality Among the Elderly in Rural China and Influencing Factors: Evidence from the Chinese Longitudinal Healthy Longevity Survey. Int. J. Environ. Res. Public Health 2019, 16, 4018. [Google Scholar] [CrossRef] [Green Version]
- Cheung, G.; Clair, V.W.-S.; Chacko, E.; Barak, Y. Financial difficulty and biopsychosocial predictors of loneliness: A cross-sectional study of community dwelling older adults. Arch. Gerontol. Geriatr. 2019, 85, 103935. [Google Scholar] [CrossRef]
- Leitch, S.; Glue, P.; Gray, A.; Greco, P.; Barak, Y. Comparison of Psychosocial Variables Associated With Loneliness in Centenarian vs. Elderly Populations in New Zealand. JAMA Netw. Open 2018, 1, e183880. [Google Scholar] [CrossRef]
- Freeman, S.; Armstrong, J.J.; Tyas, S.L.; Neufeld, E. Clinical characteristics and patterns of health deficits of centenarians receiving home care and long-term care services. Exp. Gerontol. 2017, 99, 46–52. [Google Scholar] [CrossRef]
- Jopp, D.S.; Park, M.-K.S.; Lehrfeld, J.; Paggi, M.E. Physical, cognitive, social and mental health in near-centenarians and centenarians living in New York City: Findings from the Fordham Centenarian Study. BMC Geriatr. 2016, 16, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Lv, J.; Wang, W.; Krafft, T.; Li, Y.; Zhang, F.; Yuan, F. Effects of Several Environmental Factors on Longevity and Health of the Human Population of Zhongxiang, Hubei, China. Biol. Trace Element Res. 2010, 143, 702–716. [Google Scholar] [CrossRef]
- Tedone, E.; Arosio, B.; Gussago, C.; Casati, M.; Ferri, E.; Ogliari, G.; Ronchetti, F.; Porta, A.; Massariello, F.; Nicolini, P.; et al. Leukocyte telomere length and prevalence of age-related diseases in semisupercentenarians, centenarians and centenarians’ offspring. Exp. Gerontol. 2014, 58, 90–95. [Google Scholar] [CrossRef]
- Terry, D.F.; Wilcox, M.; McCormick, M.A.; Lawler, E.; Perls, T. Cardiovascular Advantages Among the Offspring of Centenarians. J. Gerontol. Ser. A Boil. Sci. Med. Sci. 2003, 58, M425–M431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bucci, L.; Ostan, R.; Cevenini, E.; Pini, E.; Scurti, M.; Vitale, G.; Mari, D.; Caruso, C.; Sansoni, P.; Fanelli, F.; et al. Centenarians’ offspring as a model of healthy aging: A reappraisal of the data on Italian subjects and a comprehensive overview. Aging 2016, 8, 510–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Nguyen, T.; Wong, E.M.; Dugué, P.-A.; Dite, G.S.; Armstrong, N.J.; Craig, J.M.; Mather, K.A.; Sachdev, P.S.; Saffery, R.; et al. Genetic and environmental causes of variation in epigenetic aging across the lifespan. Clin. Epigenetics 2020, 12, 158. [Google Scholar] [CrossRef]
- Vaupel, J.W. Biodemography of human ageing. Nat. Cell Biol. 2010, 464, 536–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buettner, D.; Skemp, S. Blue Zones. Am. J. Lifestyle Med. 2016, 10, 318–321. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Zhou, J.; Long, A.; He, X.; Deng, X.; Chen, Y. A Comparative Study of Water Quality and Human Health Risk Assessment in Longevity Area and Adjacent Non-Longevity Area. Int. J. Environ. Res. Public Health 2019, 16, 3737. [Google Scholar] [CrossRef] [Green Version]
- Fastame, M.C.; Hitchcott, P.K.; Mulas, I.; Ruiu, M.; Penna, M.P. Resilience in Elders of the Sardinian Blue Zone: An Explorative Study. Behav. Sci. 2018, 8, 30. [Google Scholar] [CrossRef] [Green Version]
- Longo, V.D.; Antebi, A.; Bartke, A.; Barzilai, N.; Brown-Borg, H.M.; Caruso, C.; Curiel, T.J.; de Cabo, R.; Franceschi, C.; Gems, D.; et al. Interventions to Slow Aging in Humans: Are We Ready? Aging Cell 2015, 14, 497–510. [Google Scholar] [CrossRef]
- Kim, S.; Jazwinski, S.M. The Gut Microbiota and Healthy Aging: A Mini-Review. Gerontology 2018, 64, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Candela, M.; Biagi, E.; Maccaferri, S.; Turroni, S.; Brigidi, P. Intestinal microbiota is a plastic factor responding to environmental changes. Trends Microbiol. 2012, 20, 385–391. [Google Scholar] [CrossRef]
- Ticinesi, A.; Tana, C.; Nouvenne, A. The intestinal microbiome and its relevance for functionality in older persons. Curr. Opin. Clin. Nutr. Metab. Care 2019, 22, 4–12. [Google Scholar] [CrossRef]
- Loo, B.P.Y.; Lam, W.W.Y.; Mahendran, R.; Katagiri, K. How Is the Neighborhood Environment Related to the Health of Seniors Living in Hong Kong, Singapore, and Tokyo? Some Insights for Promoting Aging in Place. Ann. Am. Assoc. Geogr. 2017, 107, 812–828. [Google Scholar] [CrossRef]
- Lv, J.; Wang, W.; Li, Y. Effects of environmental factors on the longevous people in China. Arch. Gerontol. Geriatr. 2011, 53, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Deng, Q.; Wei, Y.; Zhao, Y.; Han, X.; Yin, J. Understanding the Natural and Socioeconomic Factors behind Regional Longevity in Guangxi, China: Is the Centenarian Ratio a Good Enough Indicator for Assessing the Longevity Phenomenon? Int. J. Environ. Res. Public Health 2018, 15, 938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Li, Y.; Jiang, Y.; Li, H.; Wang, W.; Yang, L. Effects of Soil Trace Elements on Longevity Population in China. Biol. Trace Element Res. 2013, 153, 119–126. [Google Scholar] [CrossRef]
- Huang, Y.; Rosenberg, M.; Hou, L.; Hu, M. Relationships among Environment, Climate, and Longevity in China. Int. J. Environ. Res. Public Health 2017, 14, 1195. [Google Scholar] [CrossRef] [Green Version]
- Favorito, J.E.; Grossl, P.R.; Davis, T.Z.; Eick, M.J.; Hankes, N. Soil-plant-animal relationships and geochemistry of selenium in the Western Phosphate Resource Area (United States): A review. Chemosphere 2021, 266, 128959. [Google Scholar] [CrossRef]
- van Bruggen, A.H.; Goss, E.M.; Havelaar, A.; van Diepeningen, A.D.; Finckh, M.; Morris, J.G. One Health-Cycling of diverse microbial communities as a connecting force for soil, plant, animal, human and ecosystem health. Sci. Total Environ. 2019, 664, 927–937. [Google Scholar] [CrossRef]
- Aßhauer, K.P.; Wemheuer, B.; Daniel, R.; Meinicke, P. Tax4Fun: Predicting functional profiles from metagenomic 16S rRNA. Bioinformatics 2015, 31, 2882–2884. [Google Scholar] [CrossRef] [PubMed]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A versatile open source tool for metagenomics. Peer J. 2016, 4, e2584. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Haas, B.J.; Gevers, D.; Earl, A.; Feldgarden, M.; Ward, D.V.; Giannoukos, G.; Ciulla, D.; Tabbaa, D.; Highlander, S.K.; Sodergren, E.; et al. Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons. Genome Res. 2011, 21, 494–504. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naive Bayesian Classifier for Rapid Assignment of rRNA Sequences into the New Bacterial Taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2012, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- Fierer, N.; Jackson, R.B. The diversity and biogeography of soil bacterial communities. Proc. Natl. Acad. Sci. USA 2006, 103, 626–631. [Google Scholar] [CrossRef] [Green Version]
- Bahram, M.; Hildebrand, F.; Forslund, S.K.; Anderson, J.L.; Soudzilovskaia, N.A.; van Bodegom, P.; Bengtsson-Palme, J.; Anslan, S.; Coelho, L.P.; Harend, H.; et al. Structure and function of the global topsoil microbiome. Nat. Cell Biol. 2018, 560, 233–237. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Wang, X.; Wen, X.; Xia, Y. Microbial community structures in different wastewater treatment plants as revealed by 454-pyrosequencing analysis. Bioresour. Technol. 2012, 117, 72–79. [Google Scholar] [CrossRef]
- Liu, Z.; Huang, S.; Sun, G.; Xu, Z.; Xu, M. Phylogenetic diversity, composition and distribution of bacterioplankton community in the Dongjiang River, China. FEMS Microbiol. Ecol. 2012, 80, 30–44. [Google Scholar] [CrossRef] [Green Version]
- Fierer, N.; Bradford, M.; Jackson, R.B. Toward An Ecological Classification Of Soil Bacteria. Ecology 2007, 88, 1354–1364. [Google Scholar] [CrossRef]
- Zoppini, A.; Amalfitano, S.; Fazi, S.; Puddu, A. Dynamics of a benthic microbial community in a riverine environment subject to hydrological fluctuations (Mulargia River, Italy). Hydrobiology 2010, 657, 37–51. [Google Scholar] [CrossRef]
- Candela, M.; Consolandi, C.; Severgnini, M.; Biagi, E.; Castiglioni, B.; Vitali, B.; De Bellis, G.; Brigidi, P. High taxonomic level fingerprint of the human intestinal microbiota by Ligase Detection Reaction-Universal Array approach. BMC Microbiol. 2010, 10, 116. [Google Scholar] [CrossRef] [Green Version]
- Shanks, O.C.; Kelty, C.A.; Archibeque, S.; Jenkins, M.; Newton, R.J.; McLellan, S.; Huse, S.M.; Sogin, M.L. Community Structures of Fecal Bacteria in Cattle from Different Animal Feeding Operations. Appl. Environ. Microbiol. 2011, 77, 2992–3001. [Google Scholar] [CrossRef] [Green Version]
- Wéry, N.; Lhoutellier, C.; Ducray, F.; Delgenès, J.-P.; Godon, J.-J. Behaviour of pathogenic and indicator bacteria during urban wastewater treatment and sludge composting, as revealed by quantitative PCR. Water Res. 2008, 42, 53–62. [Google Scholar] [CrossRef] [PubMed]
- García-Armisen, T.; Inceoğlu, Ö.; Ouattara, N.K.; Anzil, A.; Verbanck, M.A.; Brion, N.; Servais, P. Seasonal Variations and Resilience of Bacterial Communities in a Sewage Polluted Urban River. PLoS ONE 2014, 9, e92579. [Google Scholar] [CrossRef] [PubMed]
- Michaud, L.; Caruso, C.; Mangano, S.; Interdonato, F.; Bruni, V.; Giudice, A.L. Predominance ofFlavobacterium, Pseudomonas, andPolaromonaswithin the prokaryotic community of freshwater shallow lakes in the northern Victoria Land, East Antarctica. FEMS Microbiol. Ecol. 2012, 82, 391–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teeling, H.; Fuchs, B.M.; Becher, D.; Klockow, C.; Gardebrecht, A.; Bennke, C.M.; Kassabgy, M.; Huang, S.; Mann, A.J.; Waldmann, J.; et al. Substrate-Controlled Succession of Marine Bacterioplankton Populations Induced by a Phytoplankton Bloom. Science 2012, 336, 608–611. [Google Scholar] [CrossRef]
- Williams, T.J.; Wilkins, D.; Long, E.; Evans, F.; DeMaere, M.; Raftery, M.J.; Cavicchioli, R. The role of planktonicFlavobacteriain processing algal organic matter in coastal East Antarctica revealed using metagenomics and metaproteomics. Environ. Microbiol. 2012, 15, 1302–1317. [Google Scholar] [CrossRef] [PubMed]
- Zheng, G.; Yampara-Iquise, H.; Jones, J.E.; Carson, C.A. Development ofFaecalibacterium16S rRNA gene marker for identification of human faeces. J. Appl. Microbiol. 2009, 106, 634–641. [Google Scholar] [CrossRef]
- Paerl, H.W.; Gardner, W.S.; Havens, K.E.; Joyner, A.R.; McCarthy, M.J.; Newell, S.; Qin, B.; Scott, J.T. Mitigating cyanobacterial harmful algal blooms in aquatic ecosystems impacted by climate change and anthropogenic nutrients. Harmful Algae 2016, 54, 213–222. [Google Scholar] [CrossRef] [Green Version]
- Ni, J.; Yu, Y.; Feng, W.; Yan, Q.; Pan, G.; Yang, B.; Zhang, X.; Li, X. Impacts of algal blooms removal by chitosan-modified soils on zooplankton community in Taihu Lake, China. J. Environ. Sci. 2010, 22, 1500–1507. [Google Scholar] [CrossRef]
- Chen, X.; He, D.; Zhou, L.; Cao, Y.; Li, Z. Influence of hydropower stations on the water microbiota in the downstream of Jinsha River, China. Peer J. 2020, 8, e9500. [Google Scholar] [CrossRef]
- El Najjar, P.; Pfaffl, M.; Ouaini, N.; Nour, A.A.; El Azzi, D. Water and sediment microbiota diversity in response to temporal variation at the outlet of the Ibrahim River (Lebanon). Environ. Monit. Assess. 2020, 192, 201. [Google Scholar] [CrossRef]
- Zeglin, L.H. Stream microbial diversity in response to environmental changes: Review and synthesis of existing research. Front. Microbiol. 2015, 6, 454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, B.; Wang, H.; Dsouza, M.; Lou, J.; He, Y.; Dai, Z.; Brookes, P.C.; Xu, J.; Gilbert, J.A. Geographic patterns of co-occurrence network topological features for soil microbiota at continental scale in eastern China. ISME J. 2016, 10, 1891–1901. [Google Scholar] [CrossRef]
- Cavaillon, J.-M.; Legout, S. Centenary of the death of Elie Metchnikoff: A visionary and an outstanding team leader. Microbes Infect. 2016, 18, 577–594. [Google Scholar] [CrossRef] [Green Version]
- Biagi, E.; Franceschi, C.; Rampelli, S.; Severgnini, M.; Ostan, R.; Turroni, S.; Consolandi, C.; Quercia, S.; Scurti, M.; Monti, D.; et al. Gut Microbiota and Extreme Longevity. Curr. Biol. 2016, 26, 1480–1485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noronha, M.F.; Júnior, G.L.; Gilbert, J.A.; de Oliveira, V.M. Taxonomic and functional patterns across soil microbial communities of global biomes. Sci. Total Environ. 2017, 609, 1064–1074. [Google Scholar] [CrossRef] [PubMed]
- Kansole, M.M.R.; Lin, T.-F. Microcystin-LR Biodegradation by Bacillus sp.: Reaction Rates and Possible Genes Involved in the Degradation. Water 2016, 8, 508. [Google Scholar] [CrossRef] [Green Version]
- Pagé, A.P.; Yergeau, E.; Greer, C.W. Salix purpurea Stimulates the Expression of Specific Bacterial Xenobiotic Degradation Genes in a Soil Contaminated with Hydrocarbons. PLoS ONE 2015, 10, e0132062. [Google Scholar] [CrossRef] [Green Version]
- O’Mahony, M.M.; Dobson, A.; Barnes, J.D.; Singleton, I. The use of ozone in the remediation of polycyclic aromatic hydrocarbon contaminated soil. Chemosphere 2006, 63, 307–314. [Google Scholar] [CrossRef]
- Qian, X.; Liang, B.; Liu, X.; Liu, X.; Wang, J.; Liu, F.; Cui, B. Distribution, sources, and ecological risk assessment of polycyclic aromatic hydrocarbons in surface sediments from the Haihe River, a typical polluted urban river in Northern China. Environ. Sci. Pollut. Res. 2017, 24, 17153–17165. [Google Scholar] [CrossRef] [PubMed]
- Muangchinda, C.; Yamazoe, A.; Polrit, D.; Thoetkiattikul, H.; Mhuantong, W.; Champreda, V.; Pinyakong, O. Biodegradation of high concentrations of mixed polycyclic aromatic hydrocarbons by indigenous bacteria from a river sediment: A microcosm study and bacterial community analysis. Environ. Sci. Pollut. Res. 2016, 24, 4591–4602. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, L.; Xie, X.; Zhang, J.; Ding, Y.; Wu, Q. Bacterial Diversity and Community in Regional Water Microbiota between Different Towns in World’s Longevity Township Jiaoling, China. Diversity 2021, 13, 361. https://doi.org/10.3390/d13080361
Wu L, Xie X, Zhang J, Ding Y, Wu Q. Bacterial Diversity and Community in Regional Water Microbiota between Different Towns in World’s Longevity Township Jiaoling, China. Diversity. 2021; 13(8):361. https://doi.org/10.3390/d13080361
Chicago/Turabian StyleWu, Lei, Xinqiang Xie, Jumei Zhang, Yu Ding, and Qingping Wu. 2021. "Bacterial Diversity and Community in Regional Water Microbiota between Different Towns in World’s Longevity Township Jiaoling, China" Diversity 13, no. 8: 361. https://doi.org/10.3390/d13080361
APA StyleWu, L., Xie, X., Zhang, J., Ding, Y., & Wu, Q. (2021). Bacterial Diversity and Community in Regional Water Microbiota between Different Towns in World’s Longevity Township Jiaoling, China. Diversity, 13(8), 361. https://doi.org/10.3390/d13080361