
Comparative Study of the Genetic Diversity of Local Steppe Cattle Breeds from Russia, Kazakhstan and Kyrgyzstan by Microsatellite Analysis of Museum and Modern Samples †

 ,
,  ,
,  ,
, 
Abstract
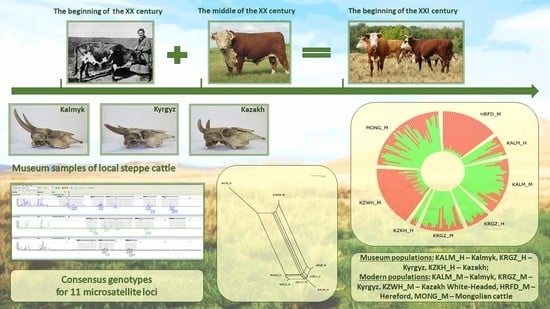
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. DNA Extraction
2.3. Microsatellite Genotyping
2.4. Data Analysis
3. Results
3.1. Estimation of Consensus Genotypes for Museum Samples
3.2. Allelic Variability and Genetic Diversity of Studied Breeds
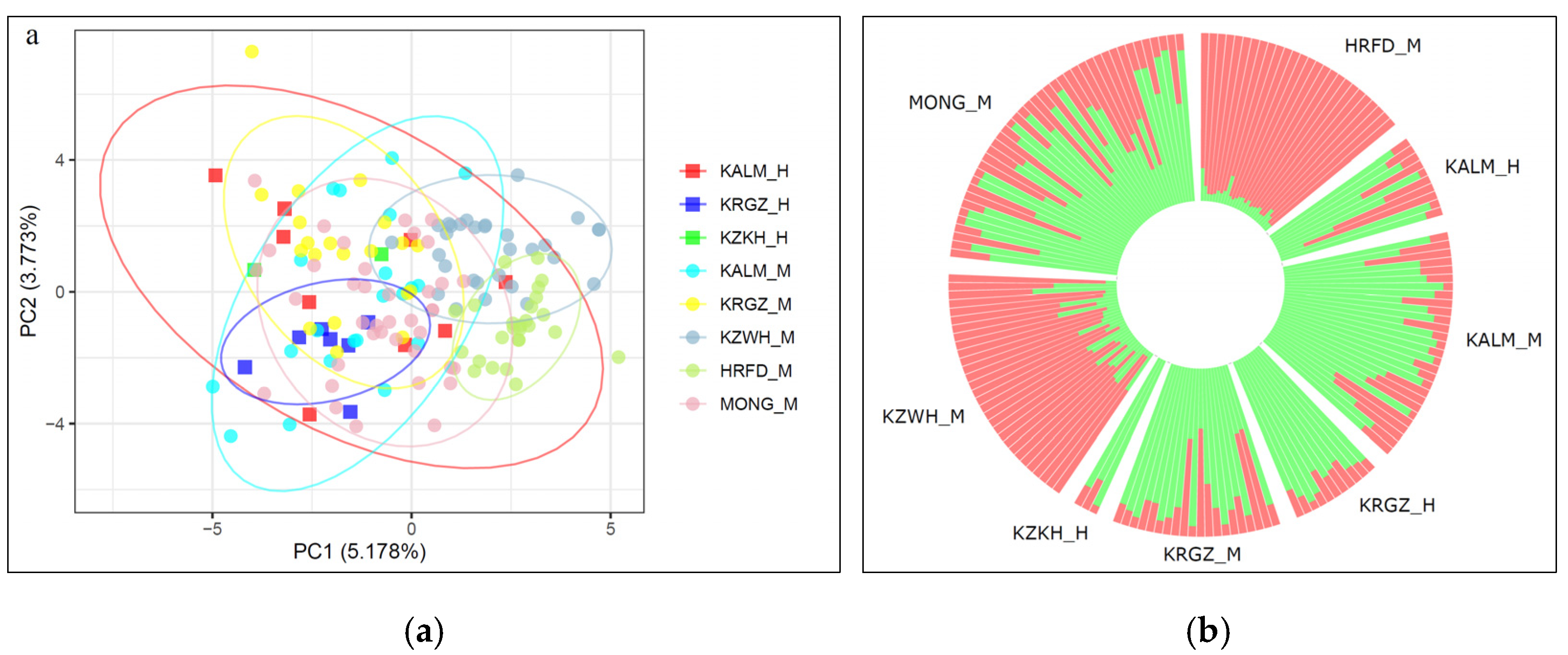
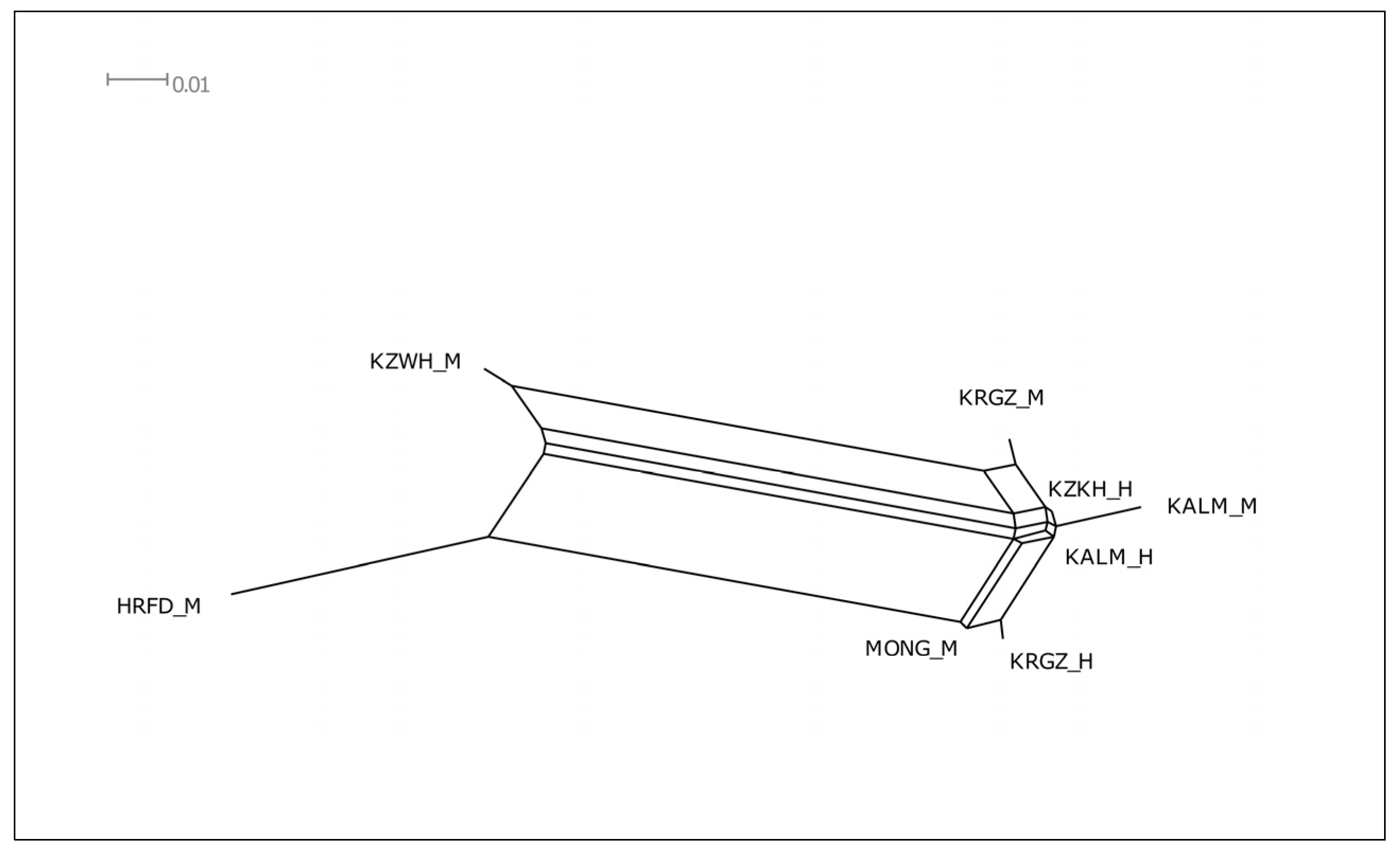
3.3. Relationships among the Studied Breeds
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jeong, C.; Wang, K.; Wilkin, S.; Taylor, W.T.T.; Miller, B.K.; Ulziibayar, S.; Stahl, R.; Chiovelli, C.; Bemmann, J.H.; Knolle, F.; et al. A Dynamic 6,000-Year Genetic History of Eurasia’s Eastern Steppe. Cell 2020, 183, 890–904.e29. [Google Scholar] [CrossRef] [PubMed]
- Wilkin, S.; Ventresca Miller, A.; Taylor, W.T.T.; Miller, B.K.; Hagan, R.W.; Bleasdale, M.; Scott, A.; Gankhuyg, S.; Ramsøe, A.; Uliziibayar, S.; et al. Dairy pastoralism sustained eastern Eurasian steppe populations for 5000 years. Nat. Ecol. Evol. 2020, 4, 346–355. [Google Scholar] [CrossRef] [PubMed]
- Nandintsetseg, B.; Shinoda, M.; Erdenetsetseg, B. Contributions of multiple climate hazards and overgrazing to the 2009/2010 winter disaster in Mongolia. Nat. Hazards 2018, 92, 109–126. [Google Scholar] [CrossRef]
- Ferret, C. Mobile pastoralism a century apart: Continuity and change in south-eastern Kazakhstan, 1910 and 2012. Cent. Asian Surv. Taylor Fr. 2018, 37, 503–525. [Google Scholar] [CrossRef]
- Suttie, J.M. Grazing Management in Mongolia; Suttie, J.M., Reynolds, S.J., Batelo, C., Eds.; Grasslands of the World, Plant Production and Protection Series 34; FAO-UN: Rome, Italy, 2005; pp. 265–304. [Google Scholar]
- Nandintsetseg, B.; Shinoda, M.; Du, C.; Munkhjargal, E. Cold-season disasters on the Eurasian steppes: Climate-driven or man-made. Sci. Rep. 2018, 8, 14769. [Google Scholar] [CrossRef]
- Leopoldov, A.V. Cattle at the First All-Union Agricultural Exhibition of 1923; Novaya Derevnya: Moscow, Russia, 1924; 178p, Available online: http://elib.cnshb.ru/books/free/0365/365927 (accessed on 15 June 2021).
- Liskun, E.F. Domestic Breeds of Cattle; State Publishing House of Agricultural Literature: Moscow, Russia, 1949; 175p. [Google Scholar]
- Ernst, L.K.; Dmitriev, N.G.; Paronyan, I.A. Genetic Resources of Farm Animals in Russia and Neighboring Countries; All-Russian Research Institute of Animal Genetics and Breeding: St. Petersburg, Russia, 1994; p. 469. [Google Scholar]
- Eldarov, N.A. About the History of Continuous Expedition Surveys in Kalmykia. Stat. Issues 2006, 6, 63–66. Available online: http://www.lib.kalmsu.ru/text/TRUD/Eldyaeva_NA/p002.pdf (accessed on 15 June 2021).
- Liskun, E.F. Russian Cattle Breeds; Novyj Agronom: Moscow, Russia, 1928; 212p. [Google Scholar]
- Pozdneev, A.M. Astrakhan Kalmyks and their attitude to Russia before the beginning of the present century. J. Minist. Public Educ. 1886, 244, 140–171. [Google Scholar]
- Lee, J.-Y. Were the Historical Oirats “Western Mongols”? An Examination of Their Uniqueness in Relation to the Mongols. Études Mongoles et Sibériennes, Centrasiatiques et Tibétaines 2016, 47. Available online: http://journals.openedition.org/emscat/2820 (accessed on 8 June 2021). [CrossRef]
- Grousset, R. The Empire of the Steppes: A History of Central Asia; Rutgers University Press: New Brunswick, NJ, USA, 1970. [Google Scholar]
- Kendirbai, G.T. The Politics of the Inner Asian Frontier and the 1771 Exodus of the Kalmyks. Inner Asia 2018, 20, 263–292. [Google Scholar] [CrossRef]
- Ivanova, O.A.; Rogov, A.L.; Fedorov, V.I. Cattle Breeding; Selkhozgiz: Moscow, Russia, 1934; 287p. [Google Scholar]
- Dmitriev, N.G.; Ernst, L.K. Animal Genetic Resources of the USSR; FAO and UNEP: Rome, Italy, 1989; Available online: http://www.fao.org/3/ah759e/AH759E07.htm (accessed on 10 February 2021).
- Sermyagin, A.A.; Dotsev, A.V.; Gladyr, E.A.; Traspov, A.A.; Deniskova, T.E.; Kostjunina, O.V.; Reyer, H.; Wimmers, K.; Barbato, M.; Paronyan, I.A.; et al. Whole-genome SNP analysis elucidates the genetic structure of Russian cattle and its relationship with Eurasian taurine breeds. Genet. Sel. Evol. 2018, 50, 37. [Google Scholar] [CrossRef]
- Yurchenko, A.; Yudin, N.; Aitnazarov, R.; Plyusnina, A.; Brukhin, V.; Soloshenko, V.; Lhasaranov, B.; Popov, R.; Paronyan, I.A.; Plemyashov, K.V.; et al. Genome-wide genotyping uncovers genetic profiles and history of the Russian cattle breeds. Heredity 2018, 120, 125–137. [Google Scholar] [CrossRef] [PubMed]
- Butusov, D.V. (Ministry of Agriculture of the Russian Federation, Moscow, Russia). Animal Genetic Resources, State Support Measures, Key Tasks. Personal communication, 2021. [Google Scholar]
- Felius, M.; Theunissen, B.; Lenstra, J. Conservation of cattle genetic resources: The role of breeds. J. Agric. Sci. 2015, 153, 152–162. [Google Scholar] [CrossRef]
- Zinovieva, N.A.; Dotsev, A.V.; Sermyagin, A.A.; Deniskova, T.E.; Abdelmanova, A.S.; Kharzinova, V.R.; Sölkner, J.; Reyer, H.; Wimmers, K.; Brem, G. Selection signatures in two oldest Russian native cattle breeds revealed using high-density single nucleotide polymorphism analysis. PLoS ONE 2020, 15, e0242200. [Google Scholar] [CrossRef] [PubMed]
- Zinovieva, N.A.; Sheiko, I.P.; Dotsev, A.V.; Sheiko, R.I.; Mikhailova, M.E.; Sermyagin, A.A.; Abdelmanova, A.S.; Kharzinova, V.R.; Reyer, H.; Wimmers, K.; et al. Genome-wide SNP analysis clearly distinguished the Belarusian Red cattle from other European cattle breeds. Anim. Genet. 2021. [Google Scholar] [CrossRef] [PubMed]
- Scherf, B.D.; Pilling, D. The Second Report on the State of the World’s Animal Genetic Resources for Food and Agriculture; FAO: Rome, Italy, 2015; Available online: http://www.fao.org/3/a-i4787e/index.html (accessed on 8 June 2021).
- Sponenberg, D.P. Conserving the Genetic Diversity of Domesticated Livestock. Diversity 2020, 12, 282. [Google Scholar] [CrossRef]
- Rowe, K.C.; Singhal, S.; Macmanes, M.D.; Ayroles, J.F.; Morelli, T.L.; Rubidge, E.M.; Bi, K.; Moritz, C.C. Museum genomics: Low-cost and high-accuracy genetic data from historical specimens. Mol. Ecol. Resour. 2011, 11, 1082–1092. [Google Scholar] [CrossRef]
- Billerman, S.M.; Walsh, J. Historical DNA as a tool to address key questions in avian biology and evolution: A review of methods, challenges, applications, and future directions. Mol. Ecol. Resour. 2019, 19, 1115–1130. [Google Scholar] [CrossRef]
- Zinovieva, N.A.; Sermyagin, A.A.; Dotsev, A.V.; Boronetslaya, O.I.; Petrikeeva, L.V.; Abdelmanova, A.S.; Brem, G. Animal genetic resources: Developing the research of allele pool of Russian cattle breeds—Minireview. Sel’skokhozyaistvennaya Biologiya 2019, 54, 631–641. [Google Scholar] [CrossRef]
- Abdelmanova, A.S.; Kharzinova, V.R.; Volkova, V.V.; Mishina, A.I.; Dotsev, A.V.; Sermyagin, A.A.; Boronetskaya, O.I.; Petrikeeva, L.V.; Chinarov, R.Y.; Brem, G.; et al. Genetic Diversity of Historical and Modern Populations of Russian Cattle Breeds Revealed by Microsatellite Analysis. Genes 2020, 11, 940. [Google Scholar] [CrossRef]
- McHugo, G.P.; Dover, M.J.; MacHugh, D.E. Unlocking the origins and biology of domestic animals using ancient DNA and paleogenomics. BMC Biol. 2019, 17, 98. [Google Scholar] [CrossRef]
- Putman, A.I.; Carbone, I. Challenges in analysis and interpretation of microsatellite data for population genetic studies. Ecol. Evol. 2014, 4, 4399–4428. [Google Scholar] [CrossRef]
- Laoun, A.; Harkat, S.; Lafri, M.; Gaouar, S.B.S.; Belabdi, I.; Ciani, E.; De Groot, M.; Blanquet, V.; Leroy, G.; Rognon, X.; et al. Inference of Breed Structure in Farm Animals: Empirical Comparison between SNP and Microsatellite Performance. Genes 2020, 11, 57. [Google Scholar] [CrossRef]
- Eusebi, P.G.; Martinez, A.; Cortes, O. Genomic Tools for Effective Conservation of Livestock Breed Diversity. Diversity 2020, 12, 8. [Google Scholar] [CrossRef]
- Brenig, B.; Schütz, E. Recent development of allele frequencies and exclusion probabilities of microsatellites used for parentage control in the German Holstein Friesian cattle population. BMC Genet. 2016, 17, 18. [Google Scholar] [CrossRef] [PubMed]
- Molecular Genetic Characterization of Animal Genetic Resources; FAO Animal Production and Health Guidelines. FAO: Rome, Italy, 2011; pp. 68–69. Available online: http://www.fao.org/3/i2413e/i2413e00.pdf (accessed on 8 June 2021).
- Abdelmanova, A.S.; Mishina, A.I.; Volkova, V.V.; Chinarov, R.Y.; Sermyagin, A.A.; Dotsev, A.V.; Boronetskaya, O.I.; Petrikeeva, L.V.; Kostyunina, O.V.; Brem, G.; et al. Comparative study of different methods of DNA extraction from cattle bones specimens maintained in a craniological collection. Sel’skokhozyaistvennaya Biol. [Agric. Biol.] 2019, 54, 1110–1121. [Google Scholar] [CrossRef]
- Mondol, S.; Karanth, K.U.; Kumar, N.S.; Gopalaswamy, A.M.; Andheria, A.; Ramakrishnan, U. Evaluation of non-invasive genetic sampling methods for estimating tiger population size. Biol. Conserv. 2009, 142, 2350–2360. [Google Scholar] [CrossRef]
- Modi, S.; Habib, B.; Ghaskadbi, P.; Nigam, P.; Mondol, S. Standardization and validation of a panel of cross-species microsatellites to individually identify the Asiatic wild dog (Cuon alpinus). Peer J. 2019, 7, e7453-10. [Google Scholar] [CrossRef] [PubMed]
- Miquel, C.; Bellemain, E.; Poillot, C.; Bessière, J.; Durand, A.; Taberlet, P. Quality indexes to assess the reliability of genotypes in studies using noninvasive sampling and multiple-tube approach. Mol. Ecol. Notes 2006, 6, 985–988. [Google Scholar] [CrossRef]
- Broquet, T.; Petit, E. Quantifying genotyping errors in noninvasive population genetics. Mol. Ecol. 2004, 13, 3601–3608. [Google Scholar] [CrossRef]
- He, G.; Kang, H.; Guo, S.T.; Ji, W.H.; Qi, X.G.; Ren, Y.; Jin, X.L.; Li, B.G. Evaluating the reliability of microsatellite genotyping from low-quality DNA templates with a polynomial distribution model. Chin. Sci Bull 2011, 56, 2523–2530. [Google Scholar] [CrossRef][Green Version]
- Chapuis, M.-P.; Estoup, A. Microsatellite Null Alleles and Estimation of Population Differentiation. Mol. Biol. Evol. 2007, 24, 621–631. [Google Scholar] [CrossRef]
- Van Oosterhout, C.; Hutchinson, W.F.; Wills, D.P.M.; Shipley, P. micro-checker: Software for identifying and correcting genotyping errors in microsatellite data. Mol. Ecol. Notes 2004, 4, 535–538. [Google Scholar] [CrossRef]
- Peakall, R.; Smouse, P.E. GenAlEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research--an update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef] [PubMed]
- Keenan, K.; McGinnity, P.; Cross, T.F.; Crozier, W.W.; Prodöhl, P.A. diveRsity : An R package for the estimation and exploration of population genetics parameters and their associated errors. Methods Ecol. Evol. 2013, 4, 782–788. [Google Scholar] [CrossRef]
- Jombart, T. adegenet: A R package for the multivariate analysis of genetic markers. Bioinformatics 2008, 24, 1403–1405. [Google Scholar] [CrossRef] [PubMed]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Use R! Springer: New York, NY, USA, 2009; ISBN 978-0-387-98140-6. [Google Scholar]
- Kalinowski, S.T. Counting Alleles with Rarefaction: Private Alleles and Hierarchical Sampling Designs. Conserv. Genet. 2004, 5, 539–543. [Google Scholar] [CrossRef]
- Wilcoxon, F. Individual Comparisons by Ranking Methods. Biom. Bull. 1945, 1, 80–83. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria; Available online: https://www.R-project.org/ (accessed on 12 May 2020).
- Jost, L. G ST and its relatives do not measure differentiation. Mol. Ecol. 2008, 17, 4015–4026. [Google Scholar] [CrossRef]
- Weir, B.S.; Cockerham, C.C. Estimating F-Statistics for the Analysis of Population Structure. Evolution 1984, 38, 1358–1370. [Google Scholar] [CrossRef]
- Huson, D.H.; Bryant, D. Application of Phylogenetic Networks in Evolutionary Studies. Mol. Biol. Evol. 2006, 23, 254–267. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef]
- Kopelman, N.M.; Mayzel, J.; Jakobsson, M.; Rosenberg, N.A.; Mayrose, I. CLUMPAK: A program for identifying clustering modes and packaging population structure inferences across K. Mol. Ecol. Resour. 2015, 15, 1179–1191. [Google Scholar] [CrossRef]
- Pickrell, J.K.; Pritchard, J.K. Inference of population splits and mixtures from genome-wide allele frequency data. PLoS Genet. 2012, 8, e100296. [Google Scholar] [CrossRef]
- Fitak, R.R. optM: An R package to optimize the number of migration edges using threshold models. J. Hered. 2019. Available online: https://cran.r-project.org/web/packages/OptM/index.html (accessed on 21 June 2021).
- Hoffmann, I. Climate change and the characterization, breeding and conservation of animal genetic resources. Anim. Genet. 2010, 41, 32–46. [Google Scholar] [CrossRef] [PubMed]
- FAO. Status and trends of Animal Genetic Resources. Commission on Genetic Resources for Food and Agriculture. 2019. Available online: http://www.fao.org/3/my867en/my867en.pdf (accessed on 24 June 2021).
- Hanotte, O.; Toll, J.; Iniguez, L.; Rege, E. Farm animal genetic resources: Why and what do we need to conserve. In Options and Strategies for the Conservation of Farm Animal Genetic Resources: Report of an International Workshop, Montpellier, France, 7–10 November 2005; Gibson, J., Gamage, S., Hanotte, O., Iniguez, L., Maillard, J.C., Rischkowsky, B., Semambo, D., Toll, J., Eds.; CGIAR System-Wide Genetic Resources Programme (SGRP): Rome, Italy; Bioversity International: Rome, Italy, 2006; p. 53. [Google Scholar]
- Cao, J.; Baumung, R.; Boettcher, P.; Scherf, B.; Besbes, B.; Leroy, G. Monitoring and Progress in the Implementation of the Global Plan of Action on Animal Genetic Resources. Sustainability 2021, 13, 775. [Google Scholar] [CrossRef]
- van Breukelen, A.E.; Doekes, H.P.; Windig, J.J.; Oldenbroek, K. Characterization of Genetic Diversity Conserved in the Gene Bank for Dutch Cattle Breeds. Diversity 2019, 11, 229. [Google Scholar] [CrossRef]
- Edwards, C.J.; Connellan, J.; Wallace, P.F.; Park, S.D.E.; McCormick, F.M.; Olsaker, I.; Eythórsdóttir, E.; MacHugh, D.E.; Bailey, J.F.; Bradley, D.G. Feasibility and utility of microsatellite markers in archaeological cattle remains from a Viking Age settlement in Dublin. Anim. Genet. 2003, 34, 410–416. [Google Scholar] [CrossRef][Green Version]
- Gargani, M.; Pariset, L.; Lenstra, J.A.; De Minicis, E.; European Cattle Genetic Diversity Consortium; Valentini, A. Microsatellite genotyping of medieval cattle from central Italy suggests an old origin of Chianina and Romagnola cattle. Front. Genet. 2015, 6, 68. [Google Scholar] [CrossRef] [PubMed]
- Svishcheva, G.; Babayan, O.; Lkhasaranov, B.; Tsendsuren, A.; Abdurasulov, A.; Stolpovsky, Y. Microsatellite diversity and phylogenetic relationships among East Eurasian Bos taurus breeds with an emphasis on rare and ancient local cattle. Animals 2020, 10, 1493. [Google Scholar] [CrossRef]
- Kayumov, F.G.; Eremenko, V.K. Kalmyk Cattle in the Conditions of South Ural and West Kazakhstan; Gazprompechat: Orenburg, Russia, 2004; 383p. [Google Scholar]
- Zhumakanov, K.T.; Abdurasulov, A.H.; Zhunushov, A.T. Conservation of Gene Pool of Agricultural Animals in Kyrgyzstan. Agric. J. 2016, 9, 50–54. Available online: https://cyberleninka.ru/article/n/sohranenie-genofonda-selskohozyaystvennyh-zhivotnyh-kyrgyzstana-problema-gosudarstvennogo-znacheniya (accessed on 26 June 2020).
- Zhang, G.X.; Wang, Z.G.; Chen, W.S.; Wu, C.X.; Han, X.; Chang, H.; Zan, L.S.; Li, R.L.; Wang, J.H.; Song, W.T.; et al. Genetic diversity and population structure of indigenous yellow cattle breeds of China using 30 microsatellite markers. Anim. Genet. 2007, 38, 550–559. [Google Scholar] [CrossRef]
- Berthouly-Salazar, C.; Thévenon, S.; Van, T.N.; Nguyen, B.T.; Pham, L.D.; Chi, C.V.; Maillard, J.C. Uncontrolled admixture and loss of genetic diversity in a local Vietnamese pig breed. Ecol. Evol. 2012, 2, 962–975. [Google Scholar] [CrossRef]
- Ba, N.V.; Arakawa, A.; Ishihara, S.; Nam, L.Q.; Thuy, T.T.T.; Dinh, N.C.; Ninh, P.H.; Cuc, N.T.K.; Kikuchi, K.; Pham, L.D.; et al. Evaluation of genetic richness among Vietnamese native pig breeds using microsatellite markers. Anim. Sci. J. 2020, 91, e13343. [Google Scholar] [CrossRef]
- McTavish, E.J.; Decker, J.E.; Schnabel, R.D.; Taylor, J.F.; Hillis, D.M. New World cattle show ancestry from multiple independent domestication events. Proc. Natl. Acad. Sci. USA 2013, 110, E1398–E1406. [Google Scholar] [CrossRef] [PubMed]
- Decker, J.E.; McKay, S.D.; Rolf, M.M.; Kim, J.; Molina Alcalá, A.; Sonstegard, T.S.; Hanotte, O.; Götherström, A.; Seabury, C.M.; Praharani, L.; et al. Worldwide Patterns of Ancestry, Divergence, and Admixture in Domesticated Cattle. PLoS Genet 2014, 10, e1004254. [Google Scholar] [CrossRef] [PubMed]
- Upadhyay, M.; Chen, W.; Lenstra, J.; Goderie, C.R.; MacHugh, D.E.; Park, S.D.; Magee, D.A.; Matassino, D.; Ciani, F.; Megens, H.J.; et al. European Cattle Genetic Diversity Consortium; RPMA Crooijmans. Genetic origin, admixture and population history of aurochs (Bos primigenius) and primitive European cattle. Heredity 2017, 118, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Verdugo, M.P.; Mullin, V.E.; Scheu, A.; Mattiangeli, V.; Daly, K.G.; Maisano Delser, P.; Hare, A.J.; Burger, J.; Collins, M.J.; Kehati, R.; et al. Ancient cattle genomics, origins, and rapid turnover in the Fertile Crescent. Science 2019, 365, 173–176. [Google Scholar] [CrossRef] [PubMed]





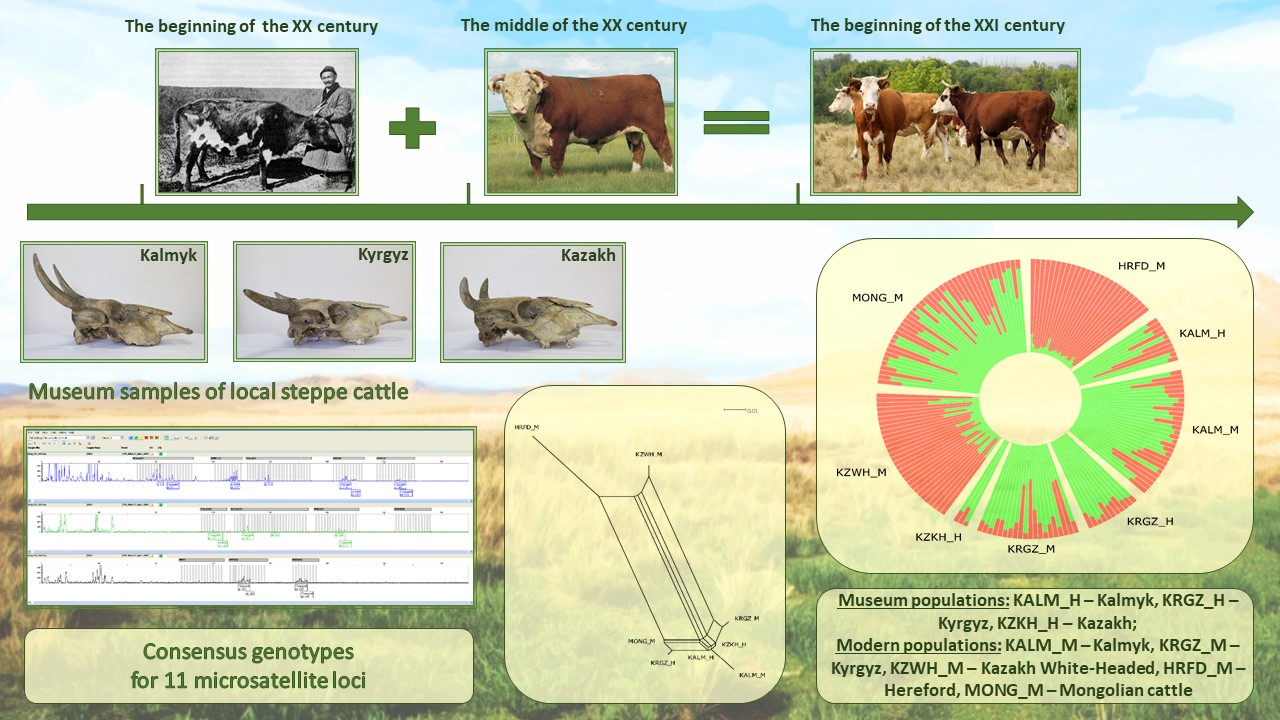{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| # | Locus | Entire Dataset | Museum Samples | Modern Samples | |||
|---|---|---|---|---|---|---|---|
| Observed Allele Ranges, bp | Number of Alleles | Observed Allele Ranges, bp | Number of Alleles | Observed Allele Ranges, bp | Number of Alleles | ||
| 1 | TGLA227 | 69–101 | 15 | 77–101 | 12 | 69–99 | 14 |
| 2 | BM2113 | 125–143 | 10 | 125–143 | 8 | 125–143 | 10 |
| 3 | TGLA53 | 152–188 | 19 | 154–184 | 13 | 152–188 | 18 |
| 4 | ETH10 | 209–225 | 8 | 213–225 | 7 | 209–225 | 8 |
| 5 | SPS115 | 244–262 | 9 | 248–260 | 5 | 244–262 | 9 |
| 6 | TGLA122 | 137–183 | 21 | 137–173 | 14 | 137–183 | 20 |
| 7 | INRA23 | 196–218 | 12 | 198–216 | 10 | 196–218 | 12 |
| 8 | TGLA126 | 107–125 | 9 | 107–123 | 7 | 111–125 | 8 |
| 9 | BM1818 | 256–274 | 10 | 256–274 | 8 | 258–274 | 9 |
| 10 | ETH225 | 140–160 | 10 | 140–158 | 7 | 140–160 | 9 |
| 11 | BM1824 | 178–190 | 7 | 178–188 | 4 | 178–190 | 7 |
| In average | 11.82 ± 1.38 | 8.63 ± 0.97 | 11.27 ± 1.30 | ||||
| Populations | KALM_H | KRGZ_H | KZKH_H | KALM_M | KRGZ_M | KZWH_M | HRFD_M | MONG_M |
|---|---|---|---|---|---|---|---|---|
| KALM_H | 72 | |||||||
| KRGZ_H | 49 | 69 | ||||||
| KZKH_H | 37 | 30 | 41 | |||||
| KALM_M | 60 | 58 | 37 | 87 | ||||
| KRGZ_M | 63 | 56 | 35 | 68 | 88 | |||
| KZWH_M | 55 | 53 | 34 | 63 | 65 | 73 | ||
| HRFD_M | 48 | 40 | 30 | 49 | 53 | 53 | 57 | |
| MONG_M | 63 | 61 | 38 | 71 | 72 | 68 | 53 | 94 |
| Population | n | Ho (M ± SE) | uHe (M ± SE) | AR (M ± SE) | uFIS (CI, 95%) |
|---|---|---|---|---|---|
| KALM_H | 10 | 0.671 ± 0.048 | 0.772 ± 0.029 * | 3.635 ± 0.199 * | 0.131 [0.033; 0.229] |
| KRGZ_H | 11 | 0.707 ± 0.053 | 0.776 ± 0.035 * | 3.693 ± 0.251 * | 0.081 [−0.055; 0.217] |
| KZKH_H | 3 | 0.818 ± 0.082 | 0.767 ± 0.067 | 3.727 ± 0.333 | −0.066 [−0.174; 0.042] |
| KALM_M | 28 | 0.736 ± 0.049 | 0.771 ± 0.034 | 3.664 ± 0.232 * | 0.044 [−0.050; 0.138] |
| KRGZ_M | 20 | 0.841 ± 0.021 | 0.778 ± 0.024 * | 3.704 ± 0.196 * | −0.085 [−0.137; −0.033] |
| KZWH_M | 30 | 0.736 ± 0.039 | 0.726 ± 0.030 | 3.336 ± 0.167 * | −0.011 [−0.063; 0.041] |
| HRFD_M | 26 | 0.668 ± 0.064 | 0.653 ± 0.053 | 2.994 ± 0.211 | −0.005 [−0.078; 0.068] |
| MONG_M | 41 | 0.672 ± 0.036 | 0.761 ± 0.020 * | 3.531 ± 0.176 * | 0.115 [0.028; 0.202] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abdelmanova, A.S.; Kharzinova, V.R.; Volkova, V.V.; Dotsev, A.V.; Sermyagin, A.A.; Boronetskaya, O.I.; Chinarov, R.Y.; Lutshikhina, E.M.; Sölkner, J.; Brem, G.; et al. Comparative Study of the Genetic Diversity of Local Steppe Cattle Breeds from Russia, Kazakhstan and Kyrgyzstan by Microsatellite Analysis of Museum and Modern Samples. Diversity 2021, 13, 351. https://doi.org/10.3390/d13080351
Abdelmanova AS, Kharzinova VR, Volkova VV, Dotsev AV, Sermyagin AA, Boronetskaya OI, Chinarov RY, Lutshikhina EM, Sölkner J, Brem G, et al. Comparative Study of the Genetic Diversity of Local Steppe Cattle Breeds from Russia, Kazakhstan and Kyrgyzstan by Microsatellite Analysis of Museum and Modern Samples. Diversity. 2021; 13(8):351. https://doi.org/10.3390/d13080351
Chicago/Turabian StyleAbdelmanova, Alexandra S., Veronika R. Kharzinova, Valeria V. Volkova, Arsen V. Dotsev, Alexander A. Sermyagin, Oksana I. Boronetskaya, Roman Yu. Chinarov, Evgeniya M. Lutshikhina, Johann Sölkner, Gottfried Brem, and et al. 2021. "Comparative Study of the Genetic Diversity of Local Steppe Cattle Breeds from Russia, Kazakhstan and Kyrgyzstan by Microsatellite Analysis of Museum and Modern Samples" Diversity 13, no. 8: 351. https://doi.org/10.3390/d13080351
APA StyleAbdelmanova, A. S., Kharzinova, V. R., Volkova, V. V., Dotsev, A. V., Sermyagin, A. A., Boronetskaya, O. I., Chinarov, R. Y., Lutshikhina, E. M., Sölkner, J., Brem, G., & Zinovieva, N. A. (2021). Comparative Study of the Genetic Diversity of Local Steppe Cattle Breeds from Russia, Kazakhstan and Kyrgyzstan by Microsatellite Analysis of Museum and Modern Samples. Diversity, 13(8), 351. https://doi.org/10.3390/d13080351