
The Phylogenetics and Biogeography of the Central Asian Hawkmoths, Hyles hippophaes and H. chamyla: Can Mitogenomics and Machine Learning Bring Clarity?

 , ,
, ,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Taxon Sampling and Processing
2.2. Species Identification and Automated Morphological Analyses
2.3. Whole Genome in Solution Capture (WISC)
2.3.1. RNA Bait Preparation
2.3.2. DNA Extractions, Library Preparation and Capturing of Museum Samples
2.4. Data Processing
2.5. Multi-Sequence Alignment of Cytochrome Oxidase Subunits
3. Results
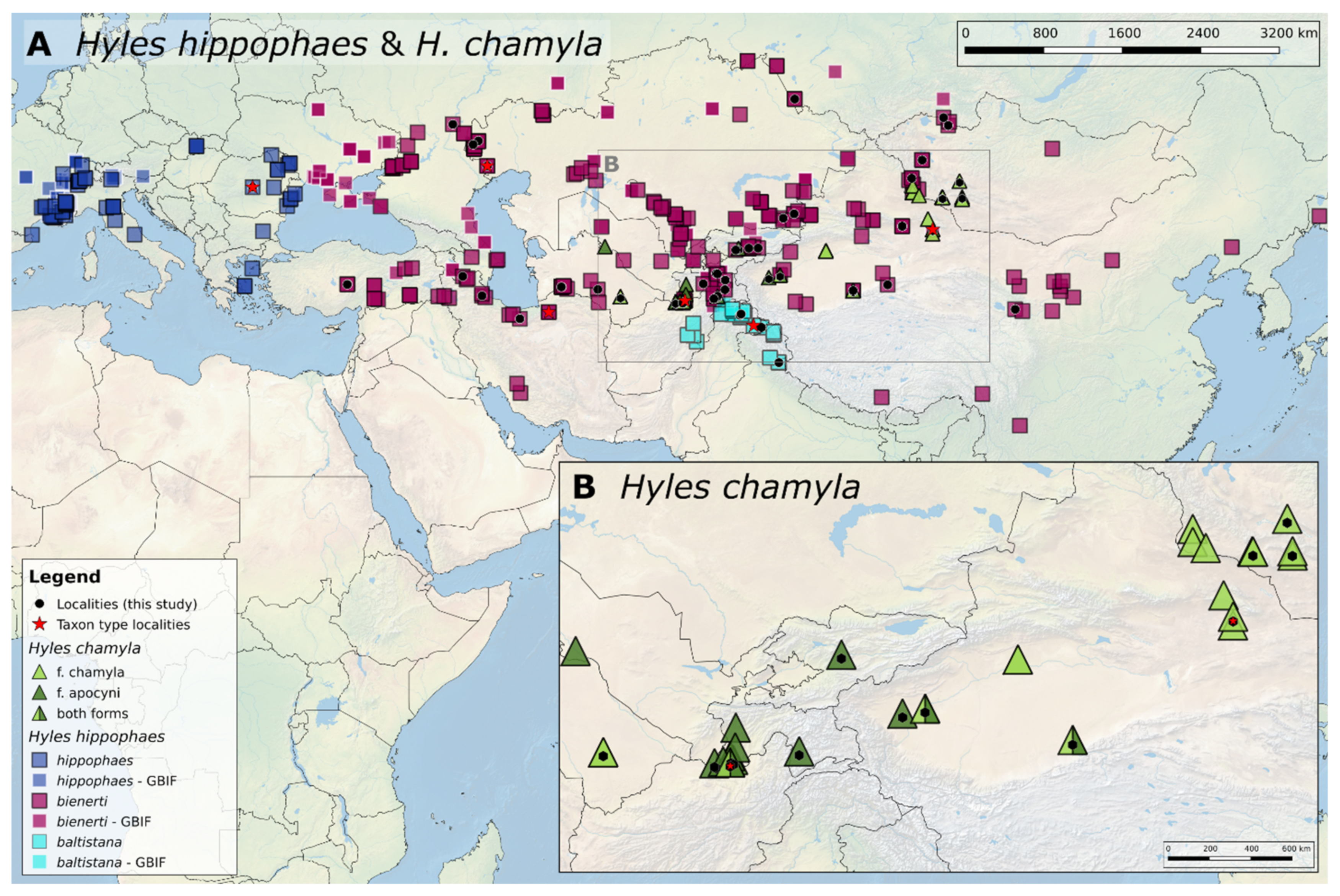
3.1. Geographic Distribution of the Hyles Hippophaes Complex
3.2. Automated Morphological Analyses
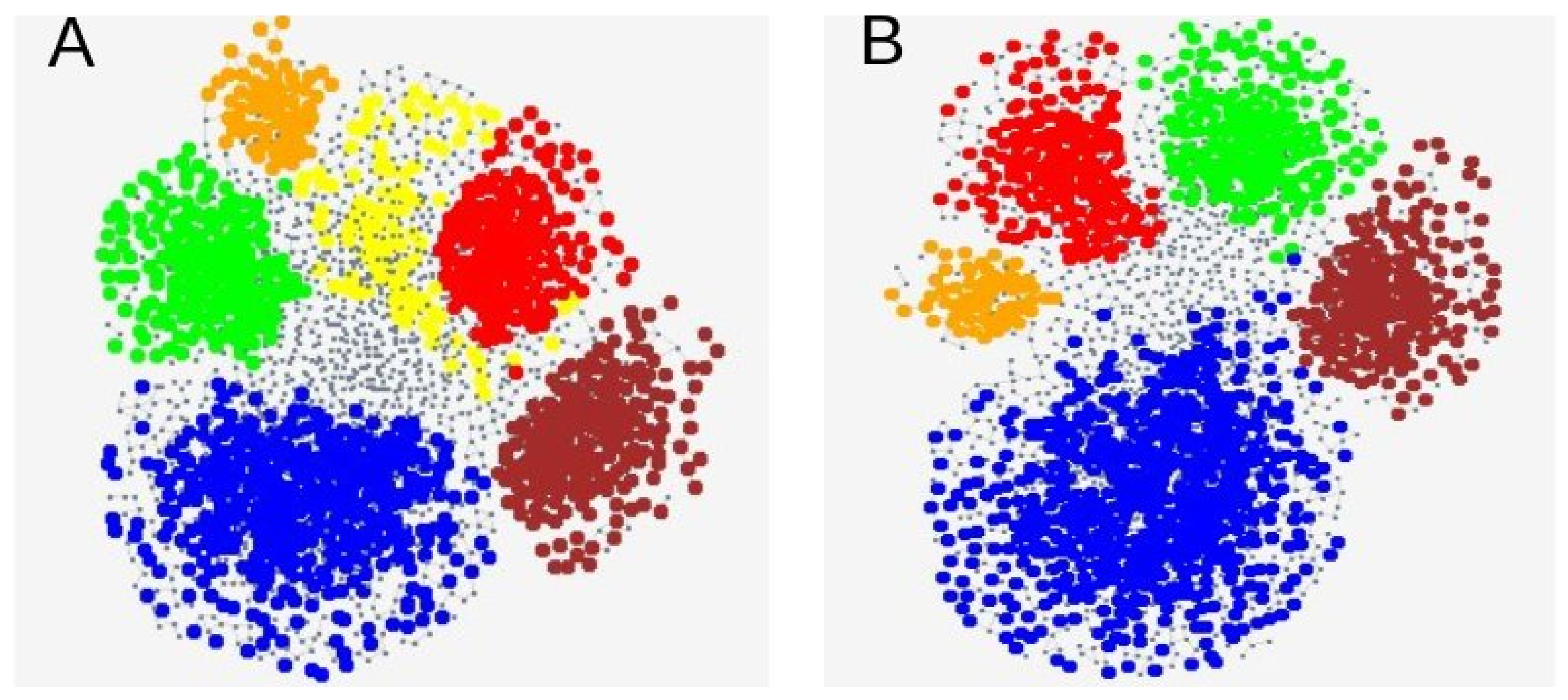
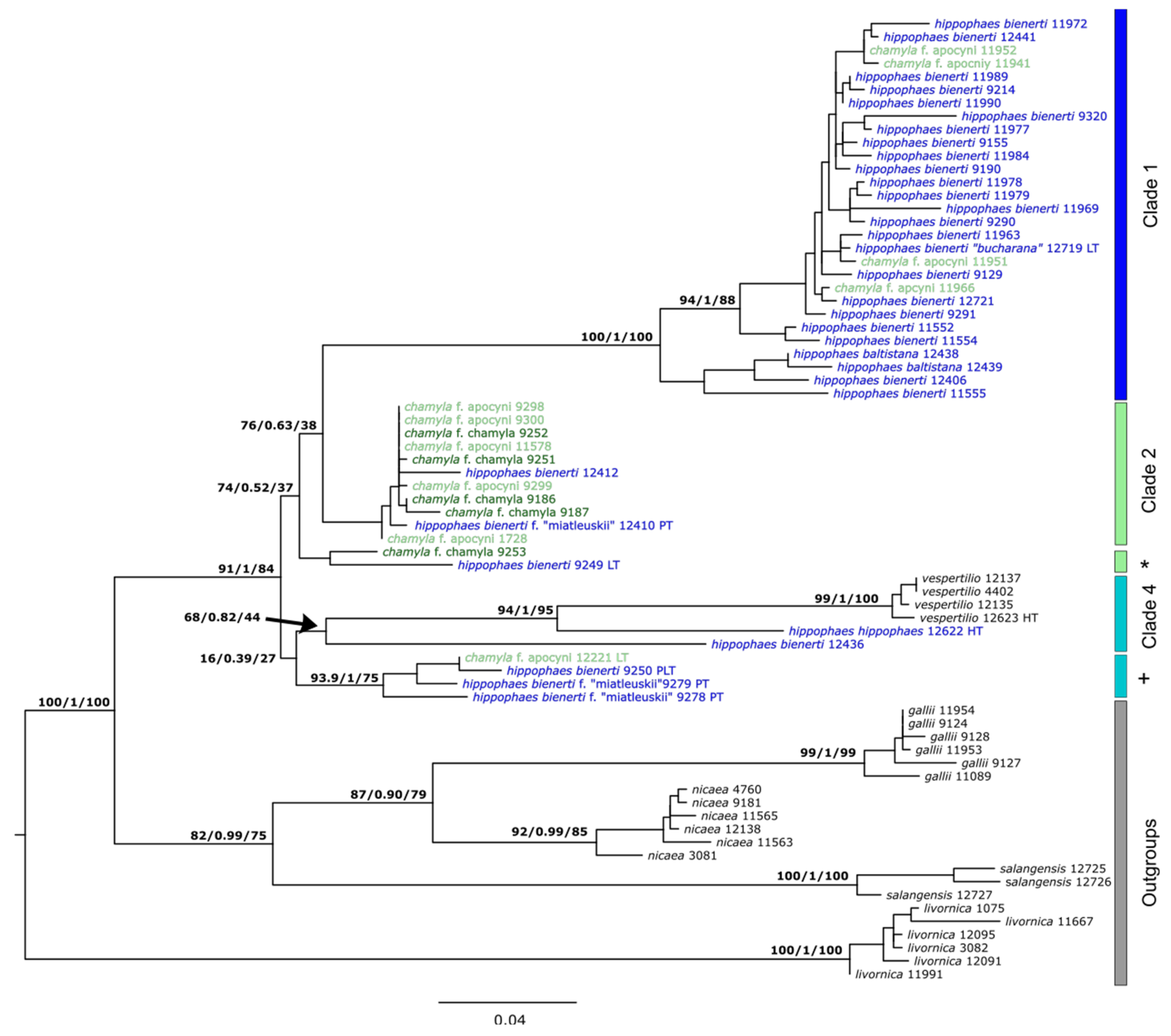
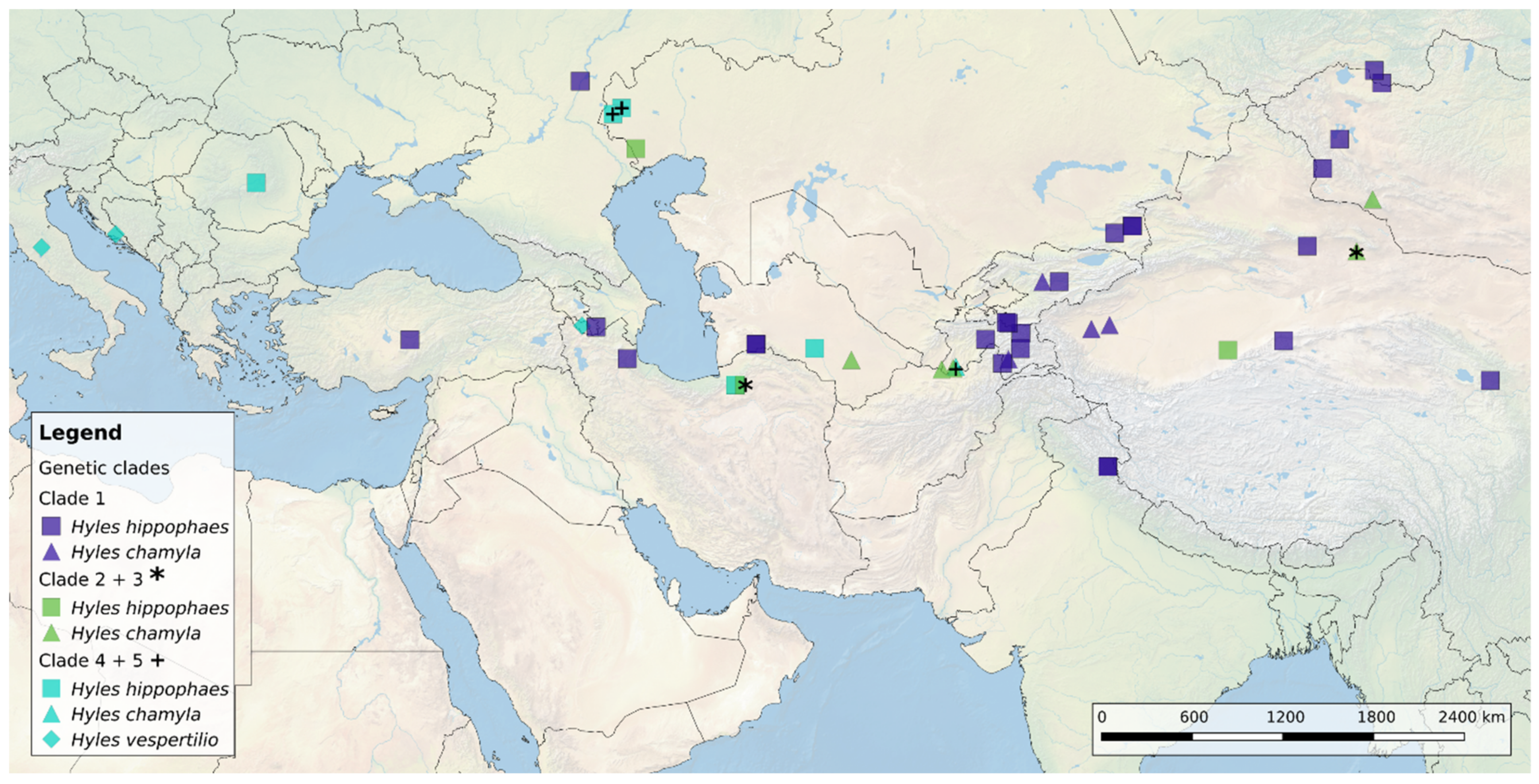
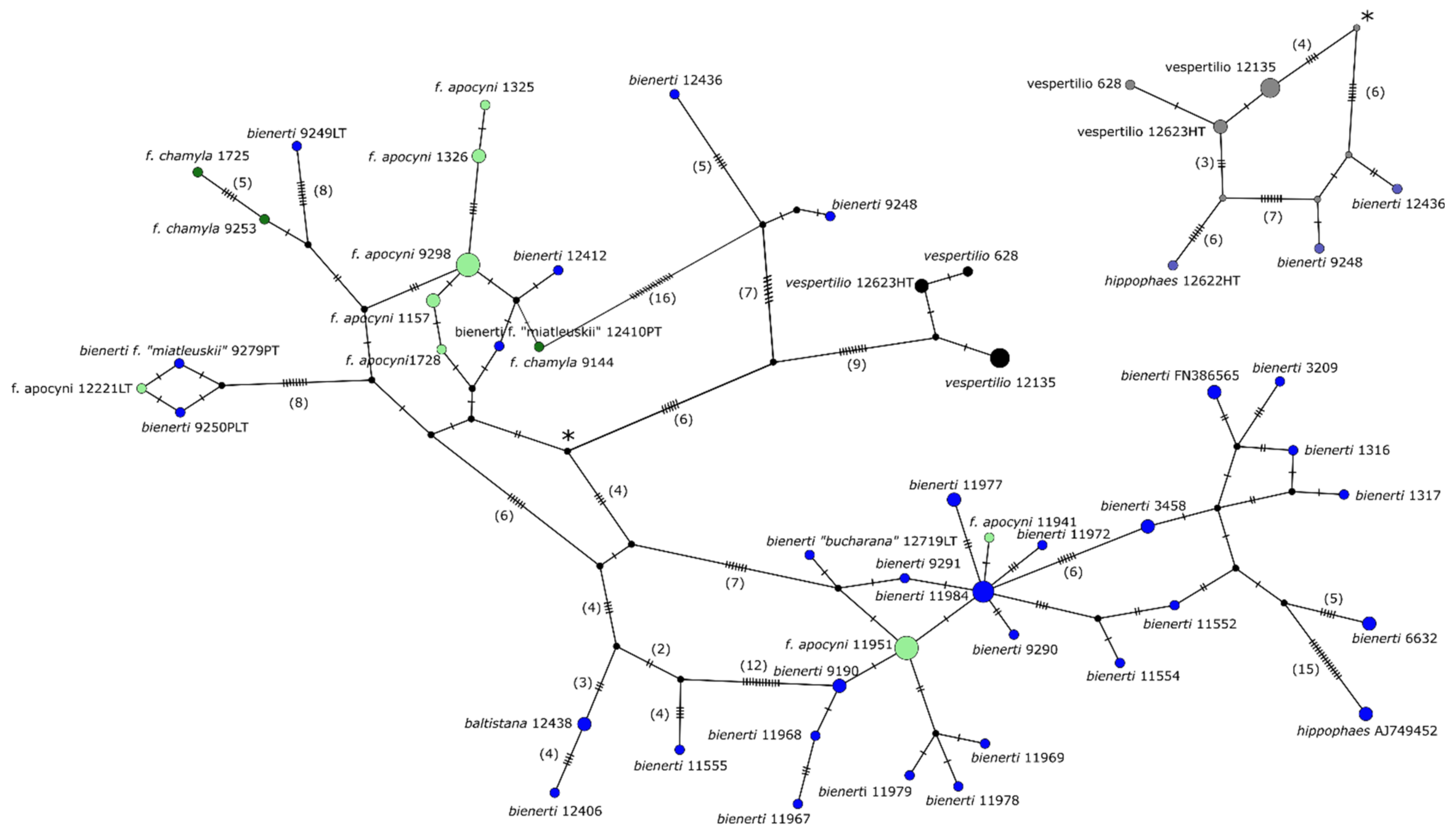
3.3. Genetic SNP Analyses
3.4. Comparison to Published Sanger Sequences
4. Discussion
4.1. Geographic Species Distribution
4.2. The Hybrid Origin of the Apocyni Phenotype
4.3. Relationship of H. hippophaes and H. vespertilio
4.4. Types and Taxonomy
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A. Morphological Species Definition of
- H. hippophaes
- H. chamyla f. chamyla
- H. chamyla f. apocyni
References
- Hundsdoerfer, A.K.; Kitching, I.J.; Wink, M. A molecular phylogeny of the hawkmoth genus Hyles (Lepidoptera: Sphingidae, Macroglossinae). Mol. Phylogenet. Evol. 2005, 35, 442–458. [Google Scholar] [CrossRef]
- Hundsdoerfer, A.K.; Lee, K.M.; Kitching, I.J.; Mutanen, M. Genome-wide SNP Data Reveal an Overestimation of Species Diversity in a Group of Hawkmoths. Genome Biol. Evol. 2019, 11, 2136–2150. [Google Scholar] [CrossRef] [PubMed]
- Hundsdoerfer, A.K.; Rubinoff, D.; Attié, M.; Wink, M.; Kitching, I.J. A revised molecular phylogeny of the globally distributed hawkmoth genus Hyles (Lepidoptera: Sphingidae), based on mitochondrial and nuclear DNA sequences. Mol. Phylogenet. Evol. 2009, 52, 852–865. [Google Scholar] [CrossRef] [PubMed]
- Kitching, I.J.; Cadiou, J.-M. Hawkmoths of the World: An Annotated and Illustrated Revisionary Checklist (Lepidoptera: Sphingidae); Cornell University Press: Ithaca, NY, USA; Natural History Museum: London, UK, 2000; ISBN 9780801437342. [Google Scholar]
- Kitching, I.J. Sphingidae Taxonomic Inventory. 2021. Available online: http://sphingidae.myspecies.info/ (accessed on 10 April 2021).
- Danner, F. Die Schwärmer der Westlichen Palaearktis: Tafelband; U. Eitschberger: Marktleuthen, Germany, 1998; ISBN 9783923807031. [Google Scholar]
- Hundsdoerfer, A.K.; Päckert, M.; Kehlmaier, C.; Strutzenberger, P.; Kitching, I.J. Museum archives revisited: Central Asiatic hawkmoths reveal exceptionally high late Pliocene species diversification (Lepidoptera, Sphingidae). Zool. Scr. 2017, 46, 552–570. [Google Scholar] [CrossRef]
- Meerman, J.C.; Smid, G. Der Hyles euphorbiae-Komplex; die Wolfsmilchschwärmer von Kreta (Lepidoptera: Sphingidae). Entomol. Z. 1998, 161–176. [Google Scholar]
- Speidel, W.; Hassler, M.; Back, H.E.; Hassler, D. Die Schmetterlingsfauna der Südlichen Sahara und Ihrer Hochgebirge Hoggar und Tassili n’Ajjer (Lepidoptera); Entomologischer Verein Apollo: Frankfurt am Main, Germany, 1989. [Google Scholar]
- Hundsdoerfer, A.K.; Mende, M.B.; Kitching, I.J.; Cordellier, M. Taxonomy, phylogeography and climate relations of the Western Palaearctic spurge hawkmoth (Lepidoptera, Sphingidae, Macroglossinae): Western Palaearctic spurge hawkmoth relations. Zool. Scr. 2011, 40, 403–417. [Google Scholar] [CrossRef]
- Denso, P. Celerio Hippophaes Esp. Dt. Ent. Z. Iris 1913, 27, 22–45. [Google Scholar]
- Shchetkin, Y.L. On the Sphingidae Fauna of Tajikistan, Report I: Towards the systematics and the biology of hawk-moths of the Kendyr (Apocynum) and the Gissar vine. Izv. Akad. Nauk Tadzhik. Ssr Otd. Estest. Vennijkh Nauk 1956, 16, 143–156. [Google Scholar]
- Zolotuhin, V.V.; Yevdoshenko, S.I. Hawk Moths (Lepidoptera: Sphingidae) of Russia and Adjacent Territories; Korporatsiya Tekhnologiy Prodvizheniya: Ulyanovsk, Russia, 2019; 480p, ISBN 978-5-94655-372-8. (In Russian) [Google Scholar]
- Pittaway, A.R. Hyles Chamyla. Available online: https://tpittaway.tripod.com/sphinx/h_cha.htm (accessed on 7 April 2021).
- The Angiosperm Phylogeny Group. An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG IV. Bot. J. Linn. Soc. 2016, 181, 1–20. [CrossRef]
- Heidel-Fischer, H.M.; Vogel, H. Molecular mechanisms of insect adaptation to plant secondary compounds. Curr. Opin. Insect. Sci. 2015, 8, 8–14. [Google Scholar] [CrossRef]
- Cappuccino, N.; Arnason, J.T. Novel chemistry of invasive exotic plants. Biol. Lett. 2006, 2, 189–193. [Google Scholar] [CrossRef] [PubMed]
- Wrońska, A.K.; Boguś, M.I.; Kaczmarek, A.; Kazek, M. Harman and norharman, metabolites of entomopathogenic fungus Conidiobolus coronatus (Entomopthorales), disorganize development of Galleria mellonella (Lepidoptera) and affect serotonin-regulating enzymes. PLoS ONE 2018, 13, e0204828. [Google Scholar] [CrossRef] [PubMed]
- Ebel, E.R.; DaCosta, J.M.; Sorenson, M.D.; Hill, R.I.; Briscoe, A.D.; Willmott, K.R.; Mullen, S.P. Rapid diversification associated with ecological specialization in Neotropical Adelpha butterflies. Mol. Ecol. 2015, 24, 2392–2405. [Google Scholar] [CrossRef] [PubMed]
- Fordyce, J.A. Host shifts and evolutionary radiations of butterflies. Proc. Biol. Sci. 2010, 277, 3735–3743. [Google Scholar] [CrossRef]
- Gilbert, M.T.P.; Moore, W.; Melchior, L.; Worobey, M. DNA extraction from dry museum beetles without conferring external morphological damage. PLoS ONE 2007, 2, e272. [Google Scholar] [CrossRef]
- Thomsen, P.F.; Elias, S.; Gilbert, M.T.P.; Haile, J.; Munch, K.; Kuzmina, S.; Froese, D.G.; Sher, A.; Holdaway, R.N.; Willerslev, E. Non-destructive sampling of ancient insect DNA. PLoS ONE 2009, 4, e5048. [Google Scholar] [CrossRef]
- Patzold, F.; Zilli, A.; Hundsdoerfer, A.K. Advantages of an easy-to-use DNA extraction method for minimal-destructive analysis of collection specimens. PLoS ONE 2020, 15, e0235222. [Google Scholar] [CrossRef]
- Watson, A.T.; O’Neill, M.A.; Kitching, I.J. Automated identification of live moths (Macrolepidoptera) using digital automated identification System (DAISY). Syst. Biodivers. 2004, 1, 287–300. [Google Scholar] [CrossRef]
- Hundsdoerfer, A.K.; Kitching, I.J. Ancient incomplete lineage sorting of Hyles and Rhodafra (Lepidoptera: Sphingidae). Org. Divers. Evol. 2020, 20, 527–536. [Google Scholar] [CrossRef]
- Gaston, K.J.; O’Neill, M.A. Automated species identification: Why not? Philos. Trans. R. Soc. Lond. B Biol. Sci. 2004, 359, 655–667. [Google Scholar] [CrossRef]
- O’Neill, M.A. DAISY: A practical tool for semi-automated species identification. Automated Taxon Identification in Systematics: Theory, Approaches and Applications; CRC Press: Boca Raton, FL, USA, 2007; Chapter 9; pp. 131–152. [Google Scholar]
- Pohl, R.; Pohl, R.F. Cognitive Illusions: A Handbook on Fallacies and Biases in Thinking, Judgement and Memory; Psychology Press: London, UK, 2004; ISBN 9781841693514. [Google Scholar]
- Stephan, K.E.; Hilgetag, C.C.; Burns, G.A.; O’Neill, M.A.; Young, M.P.; Kötter, R. Computational analysis of functional connectivity between areas of primate cerebral cortex. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2000, 355, 111–126. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, M.L.; Buenrostro, J.D.; Valdiosera, C.; Schroeder, H.; Allentoft, M.E.; Sikora, M.; Rasmussen, M.; Gravel, S.; Guillén, S.; Nekhrizov, G.; et al. Pulling out the 1%: Whole-genome capture for the targeted enrichment of ancient DNA sequencing libraries. Am. J. Hum. Genet. 2013, 93, 852–864. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, J.; Hajibabaei, M.; Blackburn, D.C.; Hanken, J.; Cantin, E.; Posfai, J.; Evans, T.C., Jr. DNA damage in preserved specimens and tissue samples: A molecular assessment. Front. Zool. 2008, 5, 18. [Google Scholar] [CrossRef]
- Gnirke, A.; Melnikov, A.; Maguire, J.; Rogov, P.; LeProust, E.M.; Brockman, W.; Fennell, T.; Giannoukos, G.; Fisher, S.; Russ, C.; et al. Solution hybrid selection with ultra-long oligonucleotides for massively parallel targeted sequencing. Nat. Biotechnol. 2009, 27, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Tin, M.M.-Y.; Economo, E.P.; Mikheyev, A.S. Sequencing degraded DNA from non-destructively sampled museum specimens for RAD-tagging and low-coverage shotgun phylogenetics. PLoS ONE 2014, 9, e96793. [Google Scholar] [CrossRef]
- Suchan, T.; Kusliy, M.A.; Khan, N.; Chauvey, L.; Tonasso-Calvière, L.; Schiavinato, S.; Southon, J.; Shapiro, B.; Keller, M.; Krause, J.; et al. Performance and automation of ancient DNA capture with RNA hyRAD probes. Submitt. Mol. Ecol. 2021. [Google Scholar]
- Pippel, M.; Jebb, D.; Patzold, F.; Winkler, S.; Vogel, H.; Myers, G.; Hiller, M.; Hundsdoerfer, A.K. A highly contiguous genome assembly of the bat hawkmoth Hyles vespertilio (Lepidoptera: Sphingidae). Gigascience 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Thornton, J. Read Recruitment Using Bowtie2 v6 (protocols.io.kevcte6). Available online: https://www.protocols.io/view/read-recruitment-using-bowtie2-kevcte6?version_warning=no (accessed on 10 April 2021).
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Picard Toolkit. Available online: https://broadinstitute.github.io/picard/ (accessed on 10 April 2021).
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef] [PubMed]
- Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 2011, 27, 2987–2993. [Google Scholar] [CrossRef] [PubMed]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed]
- vcf2phylip v2.0: Convert a VCF Matrix into Several Matrix Formats for Phylogenetic Analysis. Available online: https://github.com/edgardomortiz/vcf2phylip (accessed on 10 April 2021).
- RStudio: Integrated Development for R; RStudio, PBC: Boston, MA, USA; Available online: https://www.rstudio.com/ (accessed on 10 April 2021).
- R: A Language and Environment for Statistical Computing. Available online: https://www.r-project.org/ (accessed on 10 April 2021).
- Wickham, H.; Averick, M.; Bryan, J.; Chang, W.; McGowan, L.; François, R.; Grolemund, G.; Hayes, A.; Henry, L.; Hester, J.; et al. Welcome to the tidyverse. J. Open Source Softw. 2019, 4, 1686. [Google Scholar] [CrossRef]
- Leigh, J.W.; Bryant, D. Popart: Full-feature software for haplotype network construction. Methods Ecol. Evol. 2015, 6, 1110–1116. [Google Scholar] [CrossRef]
- Clement, M.; Snell, Q.; Walke, P.; Posada, D.; Crandall, K. TCS: Estimating gene genealogies. In Proceedings of the 16th International Parallel and Distributed Processing Symposium 2002, Ft. Lauderdale, FL, USA, 15–19 April 2001. [Google Scholar]
- Konishi, T.; Matsukuma, S.; Fuji, H.; Nakamura, D.; Satou, N.; Okano, K. Principal Component Analysis applied directly to Sequence Matrix. Sci. Rep. 2019, 9, 19297. [Google Scholar] [CrossRef]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Anisimova, M.; Gil, M.; Dufayard, J.-F.; Dessimoz, C.; Gascuel, O. Survey of branch support methods demonstrates accuracy, power, and robustness of fast likelihood-based approximation schemes. Syst. Biol. 2011, 60, 685–699. [Google Scholar] [CrossRef]
- Guindon, S.; Dufayard, J.-F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef]
- Staudinger, O. Einige neue Lepidopteren des europäischen Faunengebietes. Stettin. Entomol. Ztg. 1874, 35, 87–98. [Google Scholar]
- Erschoff, N.G. Lepidopteren von Turkestan. Stettin. Entomol. Ztg. 1874, 35, 386–417. [Google Scholar]
- Sheijuzhko, L. Zwei neue Rassen von Celerio hippophaes. Esp. Mitt. Münch. Ents. Ges. 1933, 23, 43–45. [Google Scholar]
- Bang-Haas, O. Neubeschreibungen und Berichtigungen der Exotischen Macrolepidopterenfauna. II. Entomol. Z. Frankf. Main 1939, 52, 301–302. [Google Scholar]
- Eitschberger, U.; Saldatis, A. Hyles deserticola (Staudinger, 1901) auf der Kanaren-Insel Fuerteventura (Lepidoptera, Sphingidae). Neue Entomol. Nachr. 2006, 59, 398–400. [Google Scholar]
- Marhold, K.; Lihová, J. Polyploidy, hybridization and reticulate evolution: Lessons from the Brassicaceae. Osterr. Bot. Z. 2006, 259, 143–174. [Google Scholar] [CrossRef]
- Brower, A.V.Z. Alternative facts: A reconsideration of putatively natural interspecific hybrid specimens in the genus Heliconius (Lepidoptera: Nymphalidae). Zootaxa 2018, 4499, 1–87. [Google Scholar] [CrossRef] [PubMed]
- Fusco, G.; Minelli, A. Phenotypic plasticity in development and evolution: Facts and concepts. Introduction. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2010, 365, 547–556. [Google Scholar] [CrossRef]
- Nijhout, H.F. Development and evolution of adaptive polyphenisms. Evol. Dev. 2003, 5, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Boisduval, J.B.A.E.D. Sur cinq espèces nouvelles de Lépidoptères d’Europe. Mém. Soc. Linn. 1827, 6, 109–120. [Google Scholar]
- Thevs, N.; Zerbe, S.; Kyosev, Y.; Rozi, A.; Tang, B.; Abdusalih, N.; Novitskiy, Z. Apocynum venetum L. and Apocynum pictum Schrenk (Apocynaceae) as multi-functional and multi-service plant species in Central Asia: A review on biology, ecology, and utilization. J. Appl. Bot. Food Qual. 2013, 85, 159. [Google Scholar]
- Markus, Apocynum venetum subsp, Scabrum (Russanov) in Royal Botanic Gardens, Kew (2021), The World Checklist of Vascular Plants (WCVP). Available online: https://doi.org/10.15468/6h8ucr (accessed on 1 April 2021).
- Diamond, S.E.; Kingslover, J.G. Environmental dependence of thermal reaction norms: Host plant quality can reverse the temperature-size rule. Am. Nat. 2010, 175, 1–10. [Google Scholar] [CrossRef]
- Nijhout, H.F. The control of body size in insects. Dev. Biol. 2003, 261, 1–9. [Google Scholar] [CrossRef]
- Atkinson, D. Temperature and Organism Size: A Biological Law for Ectotherms? Advances in Ecological Research; Academic Press: London, UK, 1994; Volume 25, pp. 1–58. [Google Scholar]
- Partridge, L.; Barrie, B.; Fowler, K.; French, V. Evolution and development of body size and cell size in Drosophila melanogaster in response to temperature. Evolution 1994, 48, 1269–1276. [Google Scholar] [CrossRef] [PubMed]
- Chapman, R.F.; Simpson, S.J.; Douglas, A.E. The Insects: Structure and Function; Cambridge University Press: Cambridge, UK, 2013; ISBN 9780521113892. [Google Scholar]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Patzold, F.; Marabuto, E.; Daneck, H.; O’Neill, M.A.; Kitching, I.J.; Hundsdoerfer, A.K. The Phylogenetics and Biogeography of the Central Asian Hawkmoths, Hyles hippophaes and H. chamyla: Can Mitogenomics and Machine Learning Bring Clarity? Diversity 2021, 13, 213. https://doi.org/10.3390/d13050213
Patzold F, Marabuto E, Daneck H, O’Neill MA, Kitching IJ, Hundsdoerfer AK. The Phylogenetics and Biogeography of the Central Asian Hawkmoths, Hyles hippophaes and H. chamyla: Can Mitogenomics and Machine Learning Bring Clarity? Diversity. 2021; 13(5):213. https://doi.org/10.3390/d13050213
Chicago/Turabian StylePatzold, Franziska, Eduardo Marabuto, Hana Daneck, Mark A. O’Neill, Ian J. Kitching, and Anna K. Hundsdoerfer. 2021. "The Phylogenetics and Biogeography of the Central Asian Hawkmoths, Hyles hippophaes and H. chamyla: Can Mitogenomics and Machine Learning Bring Clarity?" Diversity 13, no. 5: 213. https://doi.org/10.3390/d13050213
APA StylePatzold, F., Marabuto, E., Daneck, H., O’Neill, M. A., Kitching, I. J., & Hundsdoerfer, A. K. (2021). The Phylogenetics and Biogeography of the Central Asian Hawkmoths, Hyles hippophaes and H. chamyla: Can Mitogenomics and Machine Learning Bring Clarity? Diversity, 13(5), 213. https://doi.org/10.3390/d13050213