
HPV16-Genotyper: A Computational Tool for Risk-Assessment, Lineage Genotyping and Recombination Detection in HPV16 Sequences, Based on a Large-Scale Evolutionary Analysis


 ,
,  ,
, 
Abstract

1. Introduction
2. Materials and Methods
3. Results
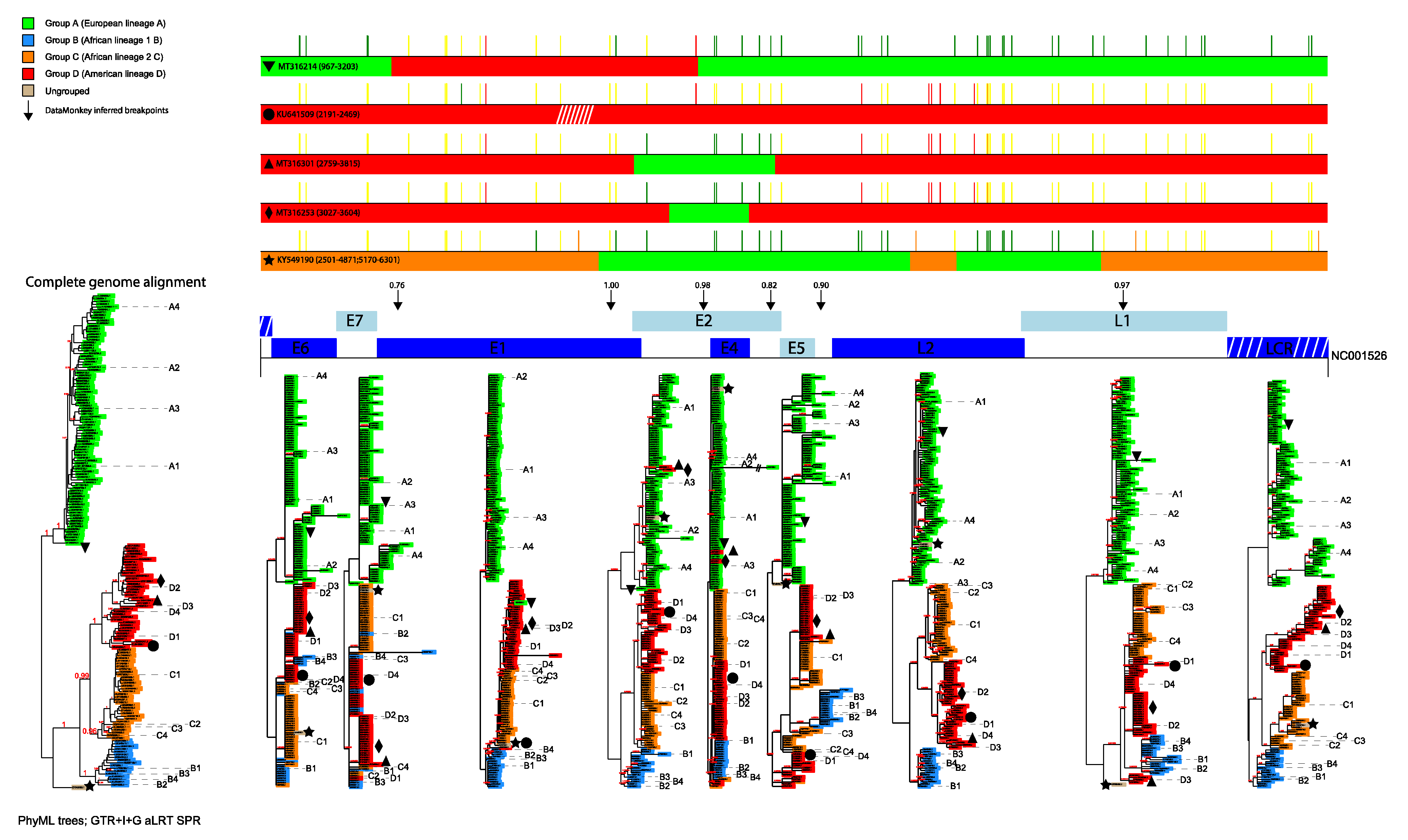
3.1. Phylogenetic Analyses
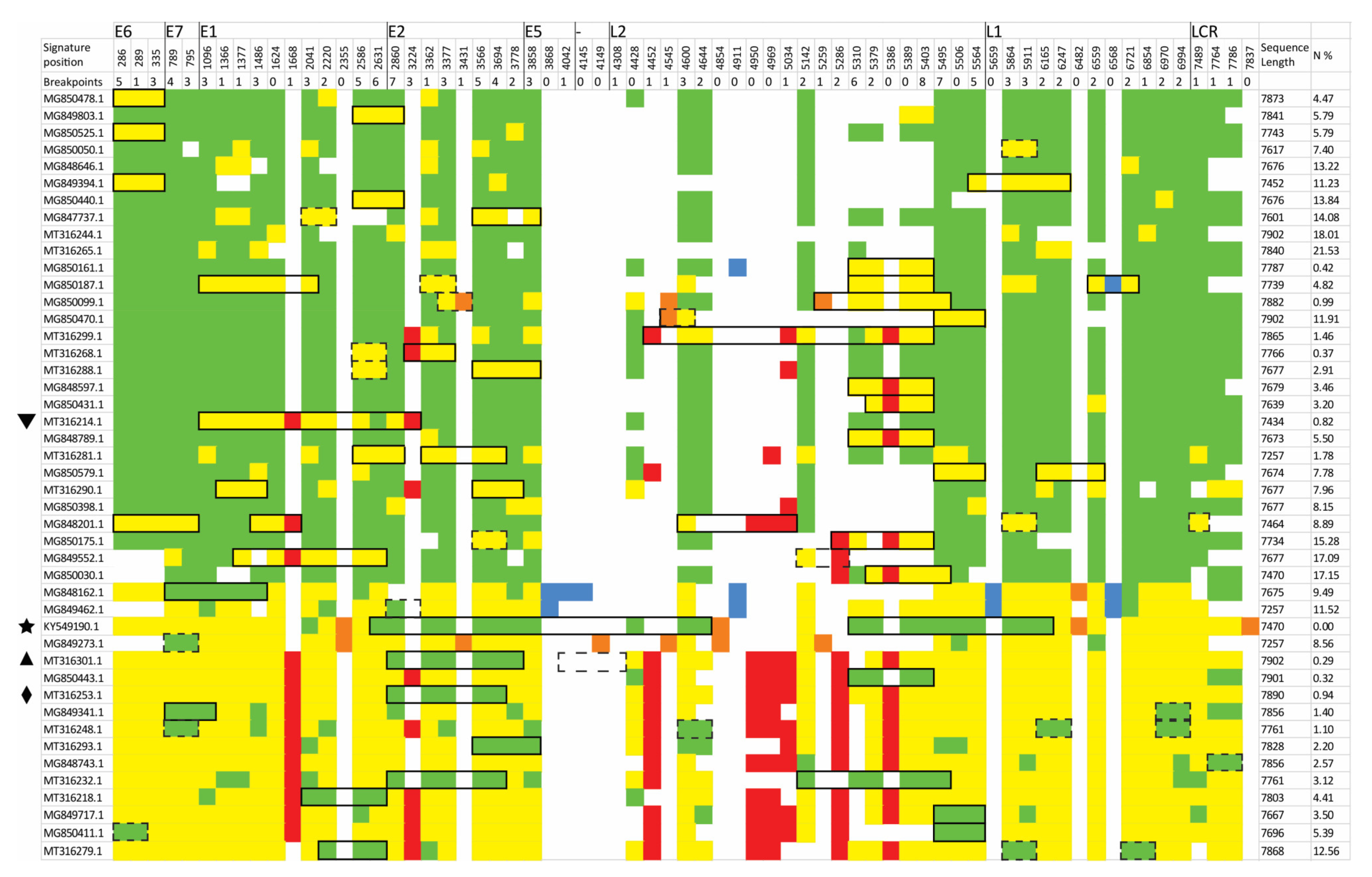
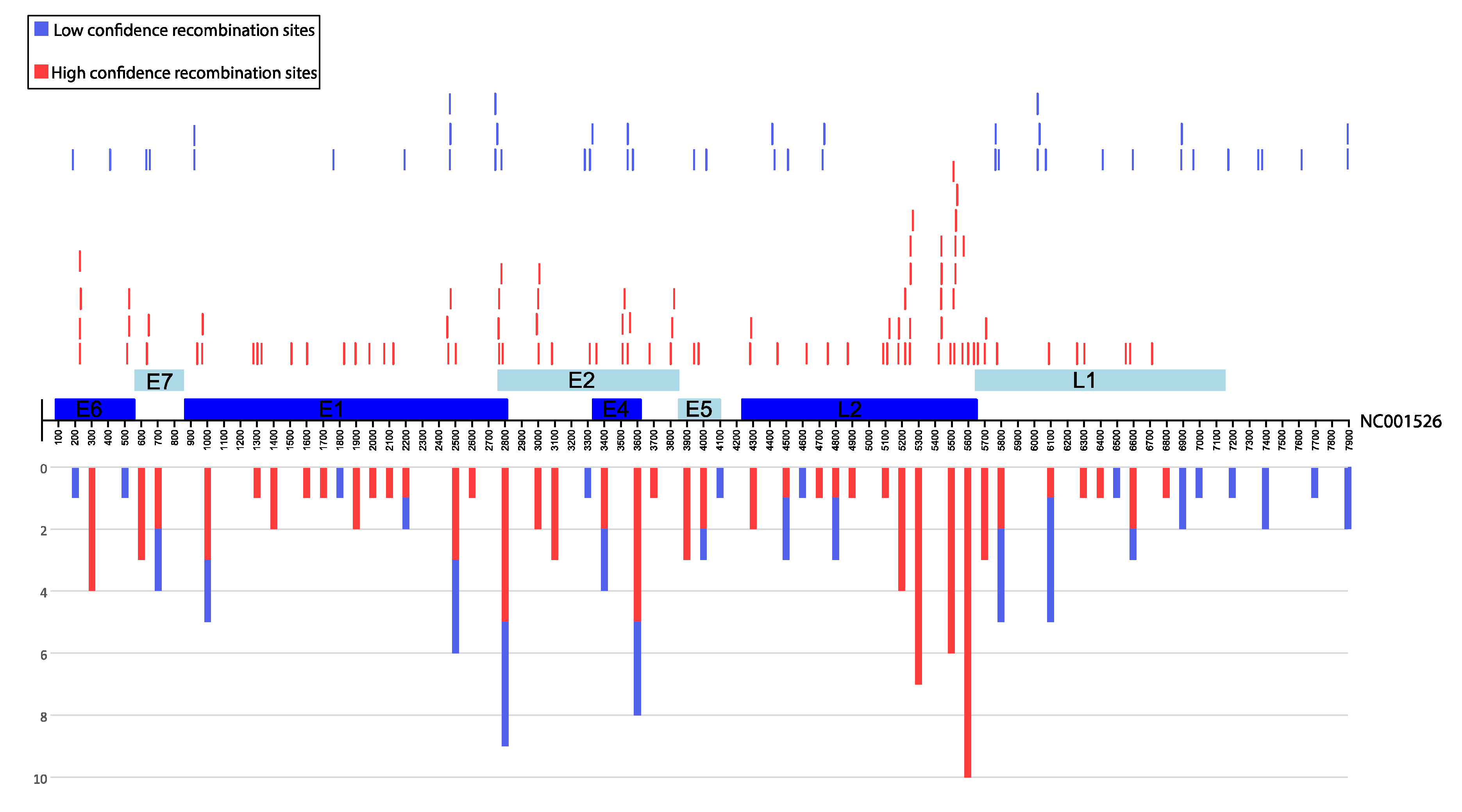
3.2. Identification of Inter-Lineage Recombinant Sequences Based on Phylogenetic Incongruence
3.3. Identification of Lineage-Specific SNPs
3.4. Rapid Detection of Inter-Lineage Recombination Events, Based on the Presence of Lineage-Specific SNPs
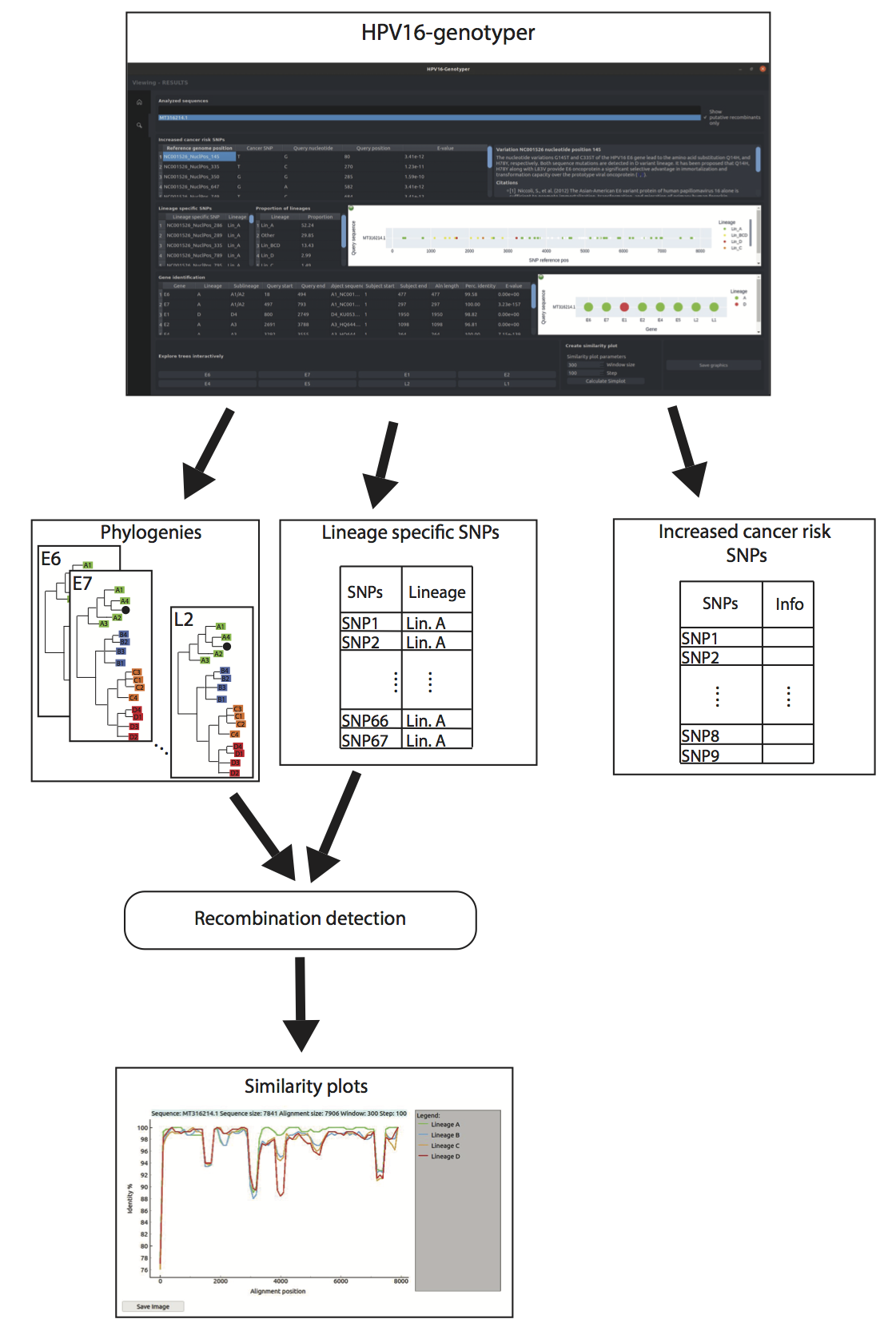
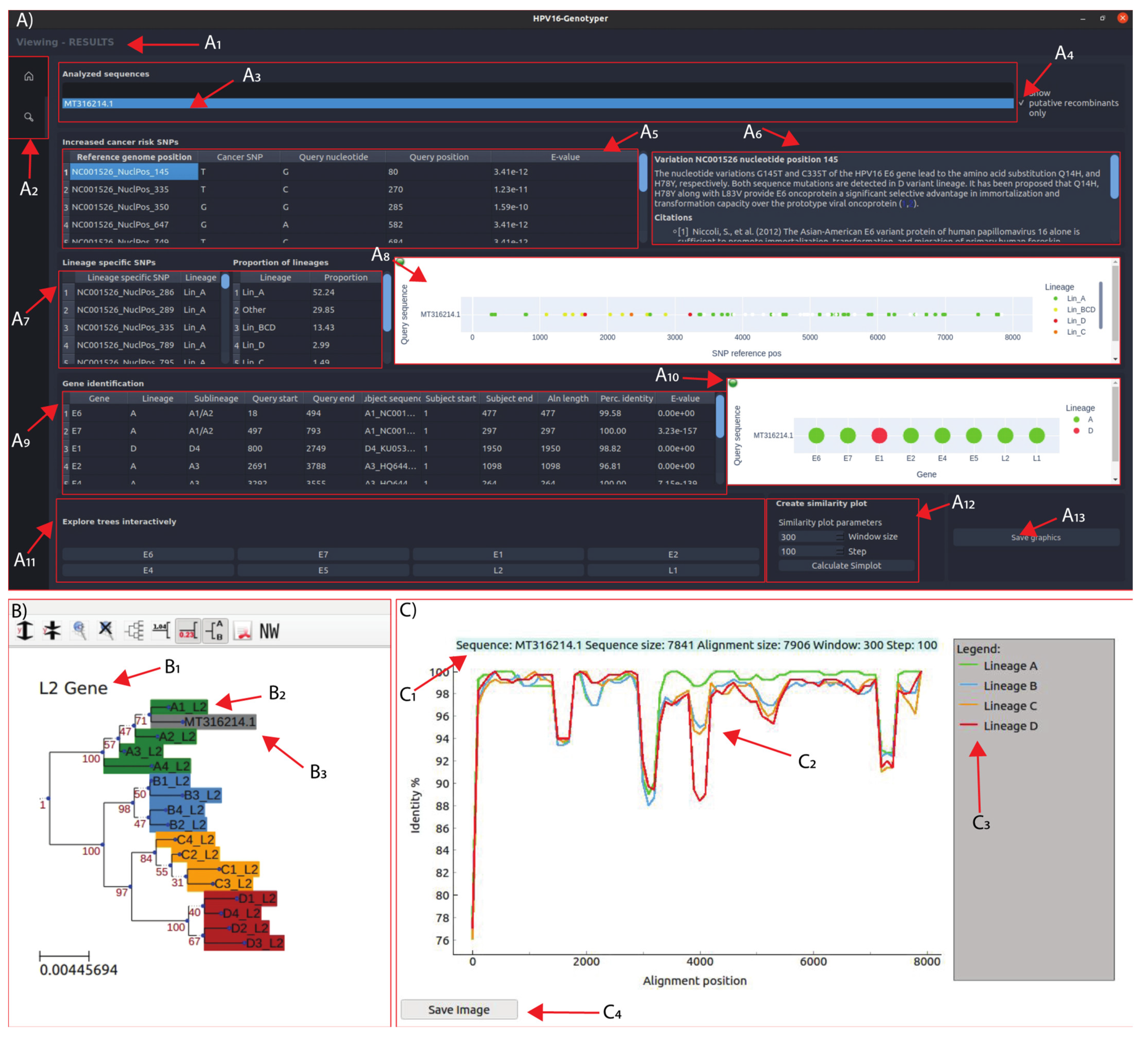
3.5. Development of the HPV16-Genotyper Computational Tool
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- zur Hausen, H. Papillomavirus Infections—A Major Cause of Human Cancers. Biochim. Biophys. Acta (BBA)—Rev. Cancer 1996, 1288, F55–F78. [Google Scholar] [CrossRef]
- Tsakogiannis, D.; Gartzonika, C.; Levidiotou-Stefanou, S.; Markoulatos, P. Molecular Approaches for HPV Genotyping and HPV-DNA Physical Status. Expert Rev. Mol. Med. 2017, 19, e1. [Google Scholar] [CrossRef]
- Bernard, H.-U.; Burk, R.D.; Chen, Z.; van Doorslaer, K.; zur Hausen, H.; de Villiers, E.-M. Classification of Papillomaviruses (PVs) Based on 189 PV Types and Proposal of Taxonomic Amendments. Virology 2010, 401, 70–79. [Google Scholar] [CrossRef] [PubMed]
- Gheit, T. Mucosal and Cutaneous Human Papillomavirus Infections and Cancer Biology. Front. Oncol. 2019, 9, 355. [Google Scholar] [CrossRef]
- Van Doorslaer, K.; Chen, Z.; Bernard, H.-U.; Chan, P.K.S.; DeSalle, R.; Dillner, J.; Forslund, O.; Haga, T.; McBride, A.A.; Villa, L.L.; et al. ICTV Virus Taxonomy Profile: Papillomaviridae. J. Gen. Virol. 2018, 99, 989–990. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Schiffman, M.; Herrero, R.; DeSalle, R.; Anastos, K.; Segondy, M.; Sahasrabuddhe, V.V.; Gravitt, P.E.; Hsing, A.W.; Burk, R.D. Evolution and Taxonomic Classification of Human Papillomavirus 16 (HPV16)-Related Variant Genomes: HPV31, HPV33, HPV35, HPV52, HPV58 and HPV67. PLoS ONE 2011, 6, e20183. [Google Scholar] [CrossRef] [PubMed]
- Mirabello, L.; Clarke, M.; Nelson, C.; Dean, M.; Wentzensen, N.; Yeager, M.; Cullen, M.; Boland, J.; NCI HPV Workshop; Schiffman, M.; et al. The Intersection of HPV Epidemiology, Genomics and Mechanistic Studies of HPV-Mediated Carcinogenesis. Viruses 2018, 10, 80. [Google Scholar] [CrossRef] [PubMed]
- McBride, A.A.; Warburton, A. The Role of Integration in Oncogenic Progression of HPV-Associated Cancers. PLoS Pathog. 2017, 13, e1006211. [Google Scholar] [CrossRef]
- Tsakogiannis, D.; Gortsilas, P.; Kyriakopoulou, Z.; Ruether, I.G.A.; Dimitriou, T.G.; Orfanoudakis, G.; Markoulatos, P. Sites of Disruption within E1 and E2 Genes of HPV16 and Association with Cervical Dysplasia: Sites of Disruption within E1 and E2 Genes of HPV16. J. Med. Virol. 2015, 87, 1973–1980. [Google Scholar] [CrossRef]
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Mathers, C.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Estimating the Global Cancer Incidence and Mortality in 2018: GLOBOCAN Sources and Methods. Int. J. Cancer 2019, 144, 1941–1953. [Google Scholar] [CrossRef]
- Li, Y.; Xu, C. Human Papillomavirus-Related Cancers. In Infectious Agents Associated Cancers: Epidemiology and Molecular Biology; Cai, Q., Yuan, Z., Lan, K., Eds.; Advances in Experimental Medicine and Biology; Springer: Singapore, 2017; pp. 23–34. ISBN 978-981-10-5765-6. [Google Scholar]
- Burk, R.D.; Harari, A.; Chen, Z. Human Papillomavirus Genome Variants. Virology 2013, 445, 232–243. [Google Scholar] [CrossRef]
- Schiffman, M.; Rodriguez, A.C.; Chen, Z.; Wacholder, S.; Herrero, R.; Hildesheim, A.; Desalle, R.; Befano, B.; Yu, K.; Safaeian, M.; et al. A Population-Based Prospective Study of Carcinogenic Human Papillomavirus Variant Lineages, Viral Persistence, and Cervical Neoplasia. Cancer Res. 2010, 70, 3159–3169. [Google Scholar] [CrossRef] [PubMed]
- Mirabello, L.; Yeager, M.; Cullen, M.; Boland, J.F.; Chen, Z.; Wentzensen, N.; Zhang, X.; Yu, K.; Yang, Q.; Mitchell, J.; et al. HPV16 Sublineage Associations With Histology-Specific Cancer Risk Using HPV Whole-Genome Sequences in 3200 Women. JNCI J. Natl. Cancer Inst. 2016, 108, djw100. [Google Scholar] [CrossRef]
- Moschonas, G.D.; Tsakogiannis, D.; Lamprou, K.A.; Mastora, E.; Dimitriou, T.G.; Kyriakopoulou, Z.; Kottaridi, C.; Karakitsos, P.; Markoulatos, P. Association of Codon 72 Polymorphism of P53 with the Severity of Cervical Dysplasia, E6-T350G and HPV16 Variant Lineages in HPV16-Infected Women. J. Med. Microbiol. 2017, 66, 1358–1365. [Google Scholar] [CrossRef]
- Tornesello, M.L.; Losito, S.; Benincasa, G.; Fulciniti, F.; Botti, G.; Greggi, S.; Buonaguro, L.; Buonaguro, F.M. Human Papillomavirus (HPV) Genotypes and HPV16 Variants and Risk of Adenocarcinoma and Squamous Cell Carcinoma of the Cervix. Gynecol. Oncol. 2011, 121, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Tsakogiannis, D.; Ruether, I.G.A.; Kyriakopoulou, Z.; Pliaka, V.; Skordas, V.; Gartzonika, C.; Levidiotou-Stefanou, S.; Markoulatos, P. Molecular and Phylogenetic Analysis of the HPV 16 E4 Gene in Cervical Lesions from Women in Greece. Arch. Virol. 2012, 157, 1729–1739. [Google Scholar] [CrossRef]
- Zacapala-Gómez, A.E.; Del Moral-Hernández, O.; Villegas-Sepúlveda, N.; Hidalgo-Miranda, A.; Romero-Córdoba, S.L.; Beltrán-Anaya, F.O.; Leyva-Vázquez, M.A.; Alarcón-Romero, L.D.C.; Illades-Aguiar, B. Changes in Global Gene Expression Profiles Induced by HPV 16 E6 Oncoprotein Variants in Cervical Carcinoma C33-A Cells. Virology 2016, 488, 187–195. [Google Scholar] [CrossRef]
- Chen, Z.; DeSalle, R.; Schiffman, M.; Herrero, R.; Wood, C.E.; Ruiz, J.C.; Clifford, G.M.; Chan, P.K.S.; Burk, R.D. Niche Adaptation and Viral Transmission of Human Papillomaviruses from Archaic Hominins to Modern Humans. PLoS Pathog. 2018, 14, e1007352. [Google Scholar] [CrossRef] [PubMed]
- Willemsen, A.; Bravo, I.G. Origin and Evolution of Papillomavirus (Onco)Genes and Genomes. Phil. Trans. R. Soc. B 2019, 374, 20180303. [Google Scholar] [CrossRef]
- Shah, S.D.; Doorbar, J.; Goldstein, R.A. Analysis of Host–Parasite Incongruence in Papillomavirus Evolution Using Importance Sampling. Mol. Biol. Evol. 2010, 27, 1301–1314. [Google Scholar] [CrossRef]
- Van Doorslaer, K. Evolution of the Papillomaviridae. Virology 2013, 445, 11–20. [Google Scholar] [CrossRef]
- Varsani, A.; van der Walt, E.; Heath, L.; Rybicki, E.P.; Williamson, A.L.; Martin, D.P. Evidence of Ancient Papillomavirus Recombination. J. Gen. Virol. 2006, 87, 2527–2531. [Google Scholar] [CrossRef]
- Simon-Loriere, E.; Holmes, E.C. Why Do RNA Viruses Recombine? Nat. Rev. Microbiol. 2011, 9, 617–626. [Google Scholar] [CrossRef]
- Borvető, F.; Bravo, I.G.; Willemsen, A. Papillomaviruses Infecting Cetaceans Exhibit Signs of Genome Adaptation Following a Recombination Event. Virus Evol. 2020, 6, veaa038. [Google Scholar] [CrossRef]
- Robles-Sikisaka, R.; Rivera, R.; Nollens, H.H.; St Leger, J.; Durden, W.N.; Stolen, M.; Burchell, J.; Wellehan, J.F.X. Evidence of Recombination and Positive Selection in Cetacean Papillomaviruses. Virology 2012, 427, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Woolford, L.; Rector, A.; Van Ranst, M.; Ducki, A.; Bennett, M.D.; Nicholls, P.K.; Warren, K.S.; Swan, R.A.; Wilcox, G.E.; O’Hara, A.J. A Novel Virus Detected in Papillomas and Carcinomas of the Endangered Western Barred Bandicoot (Perameles bougainville) Exhibits Genomic Features of Both the Papillomaviridae and Polyomaviridae. J. Virol. 2007, 81, 13280–13290. [Google Scholar] [CrossRef]
- Murahwa, A.T.; Tshabalala, M.; Williamson, A.-L. Recombination Between High-Risk Human Papillomaviruses and Non-Human Primate Papillomaviruses: Evidence of Ancient Host Switching Among Alphapapillomaviruses. J. Mol. Evol. 2020, 88, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Angulo, M.; Carvajal-Rodríguez, A. Evidence of Recombination within Human Alpha-Papillomavirus. Virol. J. 2007, 4, 33. [Google Scholar] [CrossRef] [PubMed]
- Carvajal-Rodríguez, A. Detecting Recombination and Diversifying Selection in Human Alpha-Papillomavirus. Infect. Genet. Evol. 2008, 8, 689–692. [Google Scholar] [CrossRef]
- Jiang, M.; Xi, L.F.; Edelstein, Z.R.; Galloway, D.A.; Olsem, G.J.; Lin, W.C.-C.; Kiviat, N.B. Identification of Recombinant Human Papillomavirus Type 16 Variants. Virology 2009, 394, 8–11. [Google Scholar] [CrossRef]
- Tsakogiannis, D.; Kyriakopoulou, Z.; Amoutzias, G.; Ruether, I.G.A.; Dimitriou, T.G.; Panotopoulou, E.; Markoulatos, P. Identification of Novel E6-E7 Sequence Variants of Human Papillomavirus 16. Arch. Virol. 2013, 158, 821–828. [Google Scholar] [CrossRef] [PubMed]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and Applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef]
- Edgar, R.C. Search and Clustering Orders of Magnitude Faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple Sequence Alignment with High Accuracy and High Throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Gouy, M.; Guindon, S.; Gascuel, O. SeaView Version 4: A Multiplatform Graphical User Interface for Sequence Alignment and Phylogenetic Tree Building. Mol. Biol. Evol. 2010, 27, 221–224. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K. MAFFT: A Novel Method for Rapid Multiple Sequence Alignment Based on Fast Fourier Transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [PubMed]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. JModelTest 2: More Models, New Heuristics and Parallel Computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Gascuel, O. A Simple, Fast, and Accurate Algorithm to Estimate Large Phylogenies by Maximum Likelihood. Syst. Biol. 2003, 52, 696–704. [Google Scholar] [CrossRef]
- Chevenet, F.; Brun, C.; Bañuls, A.-L.; Jacq, B.; Christen, R. TreeDyn: Towards Dynamic Graphics and Annotations for Analyses of Trees. BMC Bioinform. 2006, 7, 439. [Google Scholar] [CrossRef]
- Tsimpidis, M.; Bachoumis, G.; Mimouli, K.; Kyriakopoulou, Z.; Robertson, D.L.; Markoulatos, P.; Amoutzias, G.D. T-RECs: Rapid and Large-Scale Detection of Recombination Events among Different Evolutionary Lineages of Viral Genomes. BMC Bioinform. 2017, 18, 13. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and Analysis of Recombination Patterns in Virus Genomes. Virus Evol. 2015, 1, vev003. [Google Scholar] [CrossRef]
- Kosakovsky Pond, S.L.; Posada, D.; Gravenor, M.B.; Woelk, C.H.; Frost, S.D.W. Automated Phylogenetic Detection of Recombination Using a Genetic Algorithm. Mol. Biol. Evol. 2006, 23, 1891–1901. [Google Scholar] [CrossRef]
- Cock, P.J.A.; Antao, T.; Chang, J.T.; Chapman, B.A.; Cox, C.J.; Dalke, A.; Friedberg, I.; Hamelryck, T.; Kauff, F.; Wilczynski, B.; et al. Biopython: Freely Available Python Tools for Computational Molecular Biology and Bioinformatics. Bioinformatics 2009, 25, 1422–1423. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, L.; White, J.A.; Birch, P.R.J.; Toth, I.K. GenomeDiagram: A Python Package for the Visualization of Large-Scale Genomic Data. Bioinformatics 2006, 22, 616–617. [Google Scholar] [CrossRef]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.A.; Clamp, M.; Barton, G.J. Jalview Version 2—A Multiple Sequence Alignment Editor and Analysis Workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef]
- Huerta-Cepas, J.; Serra, F.; Bork, P. ETE 3: Reconstruction, Analysis, and Visualization of Phylogenomic Data. Mol. Biol. Evol. 2016, 33, 1635–1638. [Google Scholar] [CrossRef] [PubMed]
- Lou, H.; Boland, J.F.; Torres-Gonzalez, E.; Albanez, A.; Zhou, W.; Steinberg, M.K.; Diaw, L.; Mitchell, J.; Roberson, D.; Cullen, M.; et al. The D2 and D3 Sublineages of Human Papilloma Virus 16-Positive Cervical Cancer in Guatemala Differ in Integration Rate and Age of Diagnosis. Cancer Res. 2020, 80, 3803–3809. [Google Scholar] [CrossRef] [PubMed]
- Mandal, P.; Bhattacharjee, B.; Sen, S.; Bhattacharya, A.; Roy Chowdhury, R.; Mondal, N.R.; Sengupta, S. Complete Genome Sequences of Eight Human Papillomavirus Type 16 Asian American and European Variant Isolates from Cervical Biopsies and Lesions in Indian Women. Genome Announc. 2016, 4, e00243-16. [Google Scholar] [CrossRef] [PubMed]
- Van der Weele, P.; Meijer, C.J.L.M.; King, A.J. Whole-Genome Sequencing and Variant Analysis of Human Papillomavirus 16 Infections. J. Virol. 2017, 91, e00844-17. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, A.; Herron, J.R.; Yamada, T.; Wheeler, C.M. Human Papillomavirus Type 16 Variant Lineages Characterized by Nucleotide Sequence Analysis of the E5 Coding Segment and the E2 Hinge Region. J. Gen. Virol. 1999, 80, 595–600. [Google Scholar] [CrossRef]
- Plesa, A.; Anton, G.; Iancu, I.V.; Diaconu, C.C.; Huica, I.; Stanescu, A.D.; Socolov, D.; Nistor, E.; Popa, E.; Stoian, M.; et al. Molecular Variants of Human Papilloma Virus 16 E2, E4, E5, E6 and E7 Genes Associated with Cervical Neoplasia in Romanian Patients. Arch. Virol. 2014, 159, 3305–3320. [Google Scholar] [CrossRef]
- Swan, D.C.; Rajeevan, M.; Tortolero-Luna, G.; Follen, M.; Tucker, R.A.; Unger, E.R. Human Papillomavirus Type 16 E2 and E6/E7 Variants. Gynecol. Oncol. 2005, 96, 695–700. [Google Scholar] [CrossRef]
- Tsakogiannis, D.; Darmis, F.; Gortsilas, P.; Ruether, I.G.A.; Kyriakopoulou, Z.; Dimitriou, T.G.; Amoutzias, G.; Markoulatos, P. Nucleotide Polymorphisms of the Human Papillomavirus 16 E1 Gene. Arch. Virol. 2014, 159, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Wheeler, C.M.; Halpern, A.L.; Stewart, A.C.; Hildesheim, A.; Jenison, S.A. Human Papillomavirus Type 16 Variant Lineages in United States Populations Characterized by Nucleotide Sequence Analysis of the E6, L2, and L1 Coding Segments. J. Virol. 1995, 69, 7743–7753. [Google Scholar] [CrossRef]
- Cuninghame, S.; Jackson, R.; Lees, S.J.; Zehbe, I. Two Common Variants of Human Papillomavirus Type 16 E6 Differentially Deregulate Sugar Metabolism and Hypoxia Signalling in Permissive Human Keratinocytes. J. Gen. Virol. 2017, 98, 2310–2319. [Google Scholar] [CrossRef] [PubMed]
- Niccoli, S.; Abraham, S.; Richard, C.; Zehbe, I. The Asian-American E6 Variant Protein of Human Papillomavirus 16 Alone Is Sufficient to Promote Immortalization, Transformation, and Migration of Primary Human Foreskin Keratinocytes. J. Virol. 2012, 86, 12384–12396. [Google Scholar] [CrossRef] [PubMed]
- Tsakogiannis, D.; Papadopoulou, A.; Kontostathi, G.; Ruether, I.G.A.; Kyriakopoulou, Z.; Dimitriou, T.G.; Orfanoudakis, G.; Markoulatos, P. Molecular and Evolutionary Analysis of HPV16 E6 and E7 Genes in Greek Women. J. Med. Microbiol. 2013, 62, 1688–1696. [Google Scholar] [CrossRef] [PubMed]
- Cornet, I.; Gheit, T.; Iannacone, M.R.; Vignat, J.; Sylla, B.S.; Del Mistro, A.; Franceschi, S.; Tommasino, M.; Clifford, G.M. HPV16 Genetic Variation and the Development of Cervical Cancer Worldwide. Br. J. Cancer 2013, 108, 240–244. [Google Scholar] [CrossRef]
- Zehbe, I.; Tachezy, R.; Mytilineos, J.; Voglino, G.; Mikyskova, I.; Delius, H.; Marongiu, A.; Gissmann, L.; Wilander, E.; Tommasino, M. Human Papillomavirus 16 E6 Polymorphisms in Cervical Lesions from Different European Populations and Their Correlation with Human Leukocyte Antigen Class II Haplotypes. Int. J. Cancer 2001, 94, 711–716. [Google Scholar] [CrossRef]
- Sichero, L.; Villa, L.L. Epidemiological and Functional Implications of Molecular Variants of Human Papillomavirus. Braz. J. Med. Biol. Res. 2006, 39, 707–717. [Google Scholar] [CrossRef]
- Grodzki, M.; Besson, G.; Clavel, C.; Arslan, A.; Franceschi, S.; Birembaut, P.; Tommasino, M.; Zehbe, I. Increased Risk for Cervical Disease Progression of French Women Infected with the Human Papillomavirus Type 16 E6-350G Variant. Cancer Epidemiol. Biomark. Prev. 2006, 15, 820–822. [Google Scholar] [CrossRef]
- Fujinaga, Y.; Okazawa, K.; Nishikawa, A.; Yamakawa, Y.; Fukushima, M.; Kato, I.; Fujinaga, K. Sequence Variation of Human Papillomavirus Type 16 E7 in Preinvasive and Invasive Cervical Neoplasias. Virus Genes 1994, 9, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.S.; Kee, S.H.; Kim, J.W.; Park, N.H.; Kang, S.B.; Chang, W.H.; Lee, H.P. Major Sequence Variants in E7 Gene of Human Papillomavirus Type 16 from Cervical Cancerous and Noncancerous Lesions of Korean Women. Gynecol. Oncol. 1997, 66, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Zhan, Q.; Guo, J.; Liu, M.; Ruan, Y.; Zhu, T.; Han, L.; Li, F. Phylogeny and Polymorphism in the E6 and E7 of Human Papillomavirus: Alpha-9 (HPV16, 31, 33, 52, 58), Alpha-5 (HPV51), Alpha-6 (HPV53, 66), Alpha-7 (HPV18, 39, 59, 68) and Alpha-10 (HPV6, 44) in Women from Shanghai. Infect. Agents Cancer 2019, 14, 38. [Google Scholar] [CrossRef]
- Zhou, Z.; Yang, H.; Yang, L.; Yao, Y.; Dai, S.; Shi, L.; Li, C.; Yang, L.; Yan, Z.; Yao, Y. Human Papillomavirus Type 16 E6 and E7 Gene Variations Associated with Cervical Cancer in a Han Chinese Population. Infect. Genet. Evol. 2019, 73, 13–20. [Google Scholar] [CrossRef]
- Eschle, D.; Dürst, M.; ter Meulen, J.; Luande, J.; Eberhardt, H.C.; Pawlita, M.; Gissmann, L. Geographical Dependence of Sequence Variation in the E7 Gene of Human Papillomavirus Type 16. J. Gen. Virol. 1992, 73 Pt 7, 1829–1832. [Google Scholar] [CrossRef] [PubMed]
- Nindl, I.; Rindfleisch, K.; Lotz, B.; Schneider, A.; Dürst, M. Uniform Distribution of HPV 16 E6 and E7 Variants in Patients with Normal Histology, Cervical Intra-Epithelial Neoplasia and Cervical Cancer. Int. J. Cancer 1999, 82, 203–207. [Google Scholar] [CrossRef]
- Stephen, A.L.; Thompson, C.H.; Tattersall, M.H.; Cossart, Y.E.; Rose, B.R. Analysis of Mutations in the URR and E6/E7 Oncogenes of HPV 16 Cervical Cancer Isolates from Central China. Int. J. Cancer 2000, 86, 695–701. [Google Scholar] [CrossRef]
- Dai, S.; Yao, Y.; Yan, Z.; Zhou, Z.; Shi, L.; Wang, X.; Sun, L.; Zhang, R.; Yao, Y. The Association of Human Papillomavirus Type 16 E2 Variations with Cervical Cancer in a Han Chinese Population. Infect. Genet. Evol. 2018, 64, 241–248. [Google Scholar] [CrossRef]
- Graham, D.A.; Herrington, C.S. HPV-16 E2 Gene Disruption and Sequence Variation in CIN 3 Lesions and Invasive Squamous Cell Carcinomas of the Cervix: Relation to Numerical Chromosome Abnormalities. Mol. Pathol. 2000, 53, 201–206. [Google Scholar] [CrossRef][Green Version]
- Tsakogiannis, D.; Ruether, I.G.A.; Kyriakopoulou, Z.; Pliaka, V.; Theoharopoulou, A.; Skordas, V.; Panotopoulou, E.; Nepka, C.; Markoulatos, P. Sequence Variation Analysis of the E2 Gene of Human Papilloma Virus Type 16 in Cervical Lesions from Women in Greece. Arch. Virol. 2012, 157, 825–832. [Google Scholar] [CrossRef] [PubMed]
- Kahla, S.; Kochbati, L.; Hammami, S.; Chanoufi, M.B.; Maalej, M.; Oueslati, R. Sequence Variation in the E2-Binding Domain of HPV16 and Biological Function Evaluation in Tunisian Cervical Cancers. Biomed. Res. Int. 2014, 2014, 639321. [Google Scholar] [CrossRef]
- Clifford, G.M.; Tenet, V.; Georges, D.; Alemany, L.; Pavón, M.A.; Chen, Z.; Yeager, M.; Cullen, M.; Boland, J.F.; Bass, S.; et al. Human Papillomavirus 16 Sub-Lineage Dispersal and Cervical Cancer Risk Worldwide: Whole Viral Genome Sequences from 7116 HPV16-Positive Women. Papillomavirus Res. 2019, 7, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Kämmer, C.; Warthorst, U.; Torrez-Martinez, N.; Wheeler, C.M.; Pfister, H. Sequence Analysis of the Long Control Region of Human Papillomavirus Type 16 Variants and Functional Consequences for P97 Promoter Activity. J. Gen. Virol. 2000, 81, 1975–1981. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NC 001526 Genomic Position | Genomic Region | Gene Position | Protein Position | A Group nt | A Group nt% | B Group nt | B Group nt% | C Group nt | C Group nt% | D Group nt | D Group nt% | AA |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 286 (T) | E6 | 204 | 68 | T | 100 | A | 100 | A | 100 | A | 100 | A |
| 289 (A) | E6 | 207 | 69 | A | 100 | G | 100 | G | 100 | G | 100 | V |
| 335 (C) | E6 | 253 | 85 | C | 98 | T | 100 | T | 100 | T | 100 | H/Y |
| 789 (T) | E7 | 228 | 76 | T | 100 | C | 100 | C | 100 | C | 100 | I |
| 795 (T) | E7 | 234 | 78 | T | 99 | G | 100 | G | 100 | G | 100 | T |
| 1096 (C) | E1 | 232 | 78 | C | 100 | G | 100 | G | 100 | G | 100 | Q/E |
| 1366 (T) | E1 | 501 | 168 | T | 99 | A | 100 | A | 100 | A | 97 | C/S |
| 1377 (C) | E1 | 512 | 171 | C | 99 | T | 100 | T | 100 | T | 97 | Y |
| 1486 (T) | E1 | 621 | 208 | T | 100 | C | 100 | C | 100 | C | 100 | L |
| 1624 (C) | E1 | 759 | 254 | C | 99 | T | 100 | T | 100 | T | 100 | L |
| 1668 (A) | E1 | 803 | 268 | A | 100 | A | 100 | A | 100 | G | 100 | A |
| 2041 (C) | E1 | 1176 | 393 | C | 100 | T | 100 | T | 100 | T | 100 | L |
| 2220 (G) | E1 | 1355 | 452 | G | 100 | C | 100 | C | 97 | C | 100 | E/D |
| 2355 (T) | E1 | 1490 | 497 | T | 100 | T | 100 | C | 100 | T | 100 | S |
| 2586 (T) | E1 | 1721 | 574 | T | 100 | C | 100 | C | 100 | C | 97 | S |
| 2631 (T) | E1 | 1766 | 589 | T | 100 | A | 100 | A | 100 | A | 100 | P |
| 2860 (C) | E2 | 105 | 35 | C | 100 | A | 100 | A | 100 | A | 100 | H/Q |
| 3224 (T) | E2 | 469 | 157 | T | 99 | T | 100 | T | 100 | A | 100 | I/L |
| 3362 (A) | E2 | 607 | 203 | A | 100 | G | 100 | G | 97 | G | 100 | N/D |
| 3362 (A) | E4 | 6 | 2 | A | 100 | G | 100 | G | 97 | G | 100 | A |
| 3377 (C) | E2 | 622 | 208 | C | 99 | G | 100 | G | 100 | G | 100 | P/A |
| 3377 (C) | E4 | 21 | 7 | C | 99 | G | 100 | G | 100 | G | 100 | L |
| 3431 (G) | E2 | 676 | 226 | G | 99 | G | 100 | A | 97 | G | 97 | T/A |
| 3431 (G) | E4 | 75 | 25 | G | 99 | G | 100 | A | 97 | G | 97 | K |
| 3566 (T) | E2 | 811 | 271 | T | 98 | G | 100 | G | 100 | G | 100 | F/V |
| 3566 (T) | E4 | 210 | 70 | T | 98 | G | 100 | G | 100 | G | 100 | H/Q |
| 3694 (T) | E2 | 939 | 313 | T | 99 | A | 100 | A | 100 | A | 100 | T |
| 3778 (G) | E2 | 1023 | 341 | G | 100 | T | 100 | T | 100 | T | 100 | W/C |
| 3858 (T) | E5 | 9 | 3 | T | 100 | C | 100 | C | 97 | C | 97 | N |
| 3868 (G) | E5 | 19 | 7 | G | 100 | A | 100 | G | 97 | G | 100 | T/A |
| 4042 (A) | E5 | 193 | 65 | G | 77 | T | 100 | G | 97 | G | 100 | L/V |
| 4145 (C) | - | 0 | 0 | C | 99 | T | 100 | C | 100 | C | 100 | - |
| 4149 (A) | - | 0 | 0 | A | 99 | A | 100 | C | 100 | A | 100 | - |
| 4308 (G) | L2 | 72 | 24 | G | 99 | A | 100 | G | 100 | G | 100 | Q |
| 4428 (G) | L2 | 192 | 64 | G | 100 | A | 100 | T | 100 | T | 97 | S |
| 4452 (T) | L2 | 216 | 72 | T | 100 | T | 100 | T | 100 | C | 100 | Y |
| 4545 (T) | L2 | 309 | 103 | T | 100 | T | 100 | G | 100 | T | 100 | P |
| 4600 (T) | L2 | 364 | 122 | T | 100 | C | 100 | C | 100 | C | 100 | S/P |
| 4644 (T) | L2 | 408 | 136 | T | 99 | A | 53 | A | 73 | A | 69 | T |
| 4854 (C) | L2 | 618 | 206 | C | 100 | C | 100 | T | 100 | C | 100 | N |
| 4911 (A) | L2 | 675 | 225 | A | 100 | T | 100 | A | 100 | A | 100 | L |
| 4950 (A) | L2 | 714 | 238 | A | 100 | A | 100 | A | 100 | G | 100 | V |
| 4969 (A) | L2 | 733 | 245 | A | 100 | A | 100 | A | 100 | G | 100 | A/T |
| 5034 (A) | L2 | 798 | 266 | A | 100 | A | 100 | A | 100 | T | 100 | F/L |
| 5142 (G) | L2 | 906 | 302 | G | 100 | A | 100 | A | 100 | A | 97 | R |
| 5259 (A) | L2 | 1023 | 341 | A | 100 | A | 100 | G | 100 | A | 100 | L |
| 5286 (T) | L2 | 1050 | 350 | T | 100 | T | 100 | T | 100 | A | 97 | T |
| 5310 (T) | L2 | 1074 | 358 | T | 100 | C | 100 | C | 100 | C | 89 | P |
| 5379 (G) | L2 | 1143 | 381 | G | 99 | A | 100 | A | 100 | A | 97 | P |
| 5386 (T) | L2 | 1150 | 384 | T | 99 | T | 100 | T | 100 | G | 97 | A/S |
| 5389 (G) | L2 | 1153 | 385 | G | 100 | A | 100 | A | 100 | A | 97 | V/I |
| 5403 (T) | L2 | 1167 | 389 | T | 99 | C | 100 | C | 100 | C | 97 | S |
| 5495 (T) | L2 | 1259 | 420 | T | 100 | C | 100 | C | 100 | C | 100 | I/T |
| 5506 (G) | L2 | 1270 | 424 | G | 100 | A | 100 | A | 100 | A | 100 | A/T |
| 5564 (C) | L2 | 1328 | 443 | C | 100 | G | 100 | G | 100 | G | 100 | A/G |
| 5659 (T) | L1 | 21 | 7 | T | 100 | C | 100 | T | 100 | T | 100 | S |
| 5864 (C) | L1 | 226 | 76 | C | 100 | T | 100 | T | 100 | T | 100 | H/Y |
| 5911 (T) | L1 | 273 | 91 | T | 100 | C | 100 | C | 100 | C | 100 | Y |
| 6165 (C) | L1 | 527 | 176 | C | 100 | A | 100 | A | 100 | A | 100 | T/N |
| 6247 (T) | L1 | 609 | 203 | T | 100 | C | 100 | C | 100 | C | 97 | T |
| 6482 (T) | L1 | 844 | 282 | T | 100 | T | 100 | C | 100 | T | 100 | P/S |
| 6559 (C) | L1 | 921 | 307 | C | 97 | T | 100 | T | 100 | T | 100 | F |
| 6568 (T) | L1 | 930 | 310 | T | 100 | A | 100 | T | 100 | T | 97 | P |
| 6721 (G) | L1 | 1083 | 361 | G | 99 | A | 100 | A | 100 | A | 100 | K |
| 6854 (C) | L1 | 1216 | 406 | C | 98 | T | 100 | T | 100 | T | 100 | L |
| 6970 (C) | L1 | 1335 | 445 | C | 100 | T | 100 | T | 100 | T | 100 | T |
| 6994 (G) | L1 | 1359 | 453 | G | 99 | A | 100 | A | 100 | A | 100 | E |
| 7489 (G) | LCR | 0 | 0 | G | 100 | A | 100 | A | 100 | A | 97 | - |
| 7764 (C) | LCR | 0 | 0 | C | 100 | T | 100 | T | 100 | T | 97 | - |
| 7786 (C) | LCR | 0 | 0 | C | 100 | T | 100 | T | 100 | T | 97 | - |
| 7837 (A) | LCR | 0 | 0 | A | 100 | A | 100 | C | 100 | A | 100 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nikolaidis, M.; Tsakogiannis, D.; Bletsa, G.; Mossialos, D.; Kottaridi, C.; Iliopoulos, I.; Markoulatos, P.; Amoutzias, G.D. HPV16-Genotyper: A Computational Tool for Risk-Assessment, Lineage Genotyping and Recombination Detection in HPV16 Sequences, Based on a Large-Scale Evolutionary Analysis. Diversity 2021, 13, 497. https://doi.org/10.3390/d13100497
Nikolaidis M, Tsakogiannis D, Bletsa G, Mossialos D, Kottaridi C, Iliopoulos I, Markoulatos P, Amoutzias GD. HPV16-Genotyper: A Computational Tool for Risk-Assessment, Lineage Genotyping and Recombination Detection in HPV16 Sequences, Based on a Large-Scale Evolutionary Analysis. Diversity. 2021; 13(10):497. https://doi.org/10.3390/d13100497
Chicago/Turabian StyleNikolaidis, Marios, Dimitris Tsakogiannis, Garyfalia Bletsa, Dimitris Mossialos, Christine Kottaridi, Ioannis Iliopoulos, Panayotis Markoulatos, and Grigoris D. Amoutzias. 2021. "HPV16-Genotyper: A Computational Tool for Risk-Assessment, Lineage Genotyping and Recombination Detection in HPV16 Sequences, Based on a Large-Scale Evolutionary Analysis" Diversity 13, no. 10: 497. https://doi.org/10.3390/d13100497
APA StyleNikolaidis, M., Tsakogiannis, D., Bletsa, G., Mossialos, D., Kottaridi, C., Iliopoulos, I., Markoulatos, P., & Amoutzias, G. D. (2021). HPV16-Genotyper: A Computational Tool for Risk-Assessment, Lineage Genotyping and Recombination Detection in HPV16 Sequences, Based on a Large-Scale Evolutionary Analysis. Diversity, 13(10), 497. https://doi.org/10.3390/d13100497