Mitochondrial Genome Diversity in Collembola: Phylogeny, Dating and Gene Order

 ,
,  ,
, 

Abstract
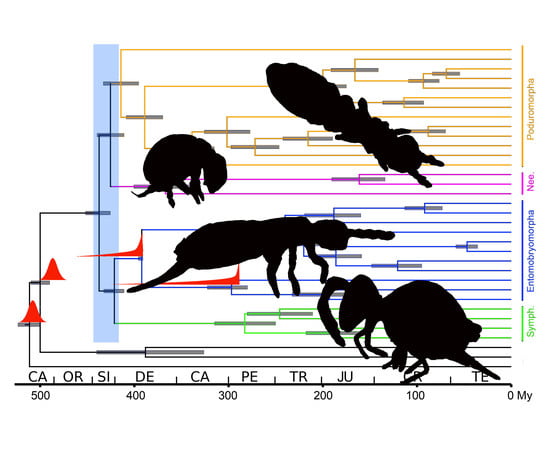
1. Introduction
2. Materials and Methods
2.1. Sequencing of the Mt-genomes of Dicyrtomina saundersi and Neelus murinus
2.2. Phylogenetic Analysis
2.3. Dating Analysis
2.4. Gene Order
3. Results
3.1. Description of Two New Genomes
3.2. Data Set
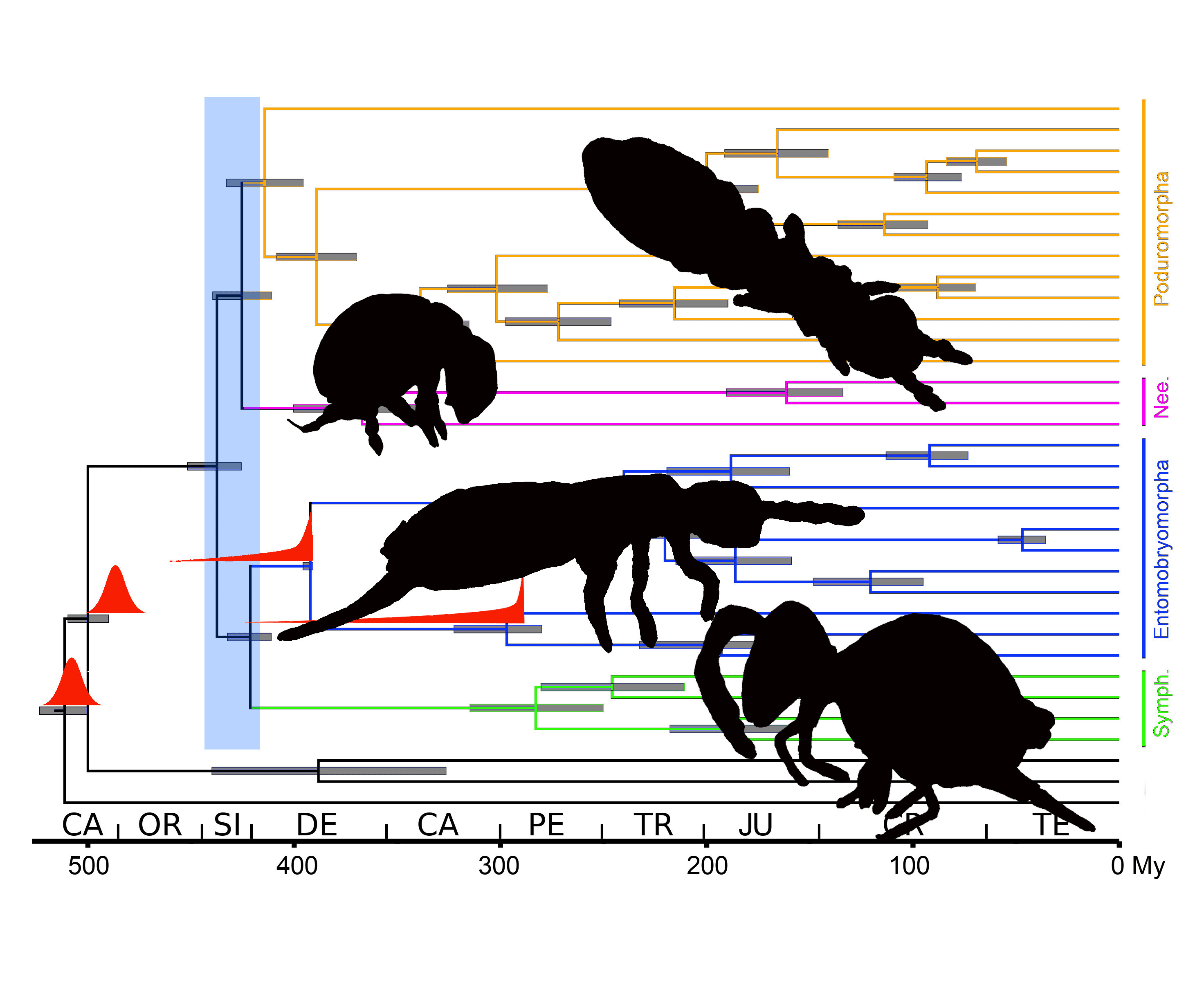
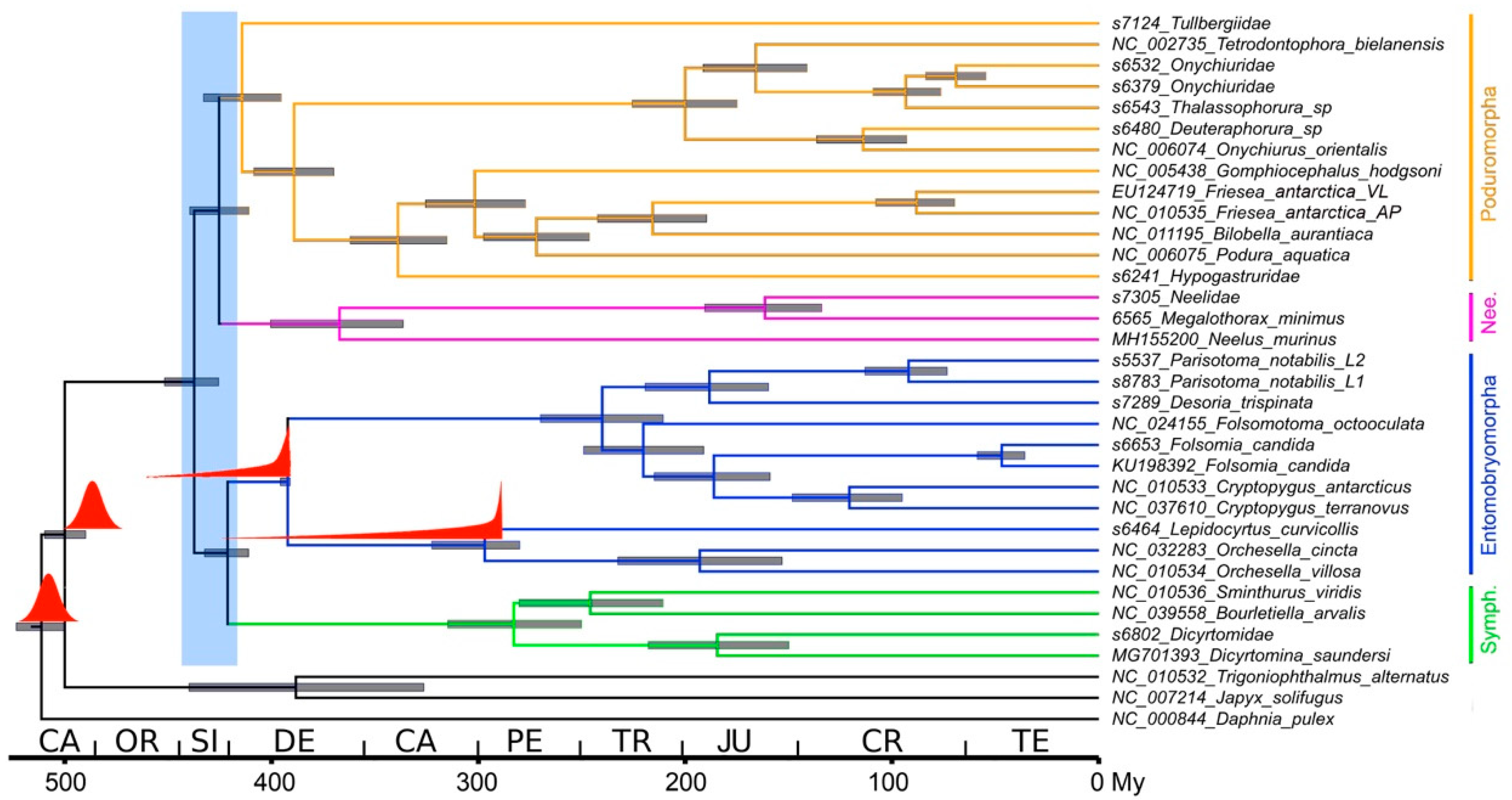
3.3. Phylogenetic Analysis
3.4. Dating Analysis
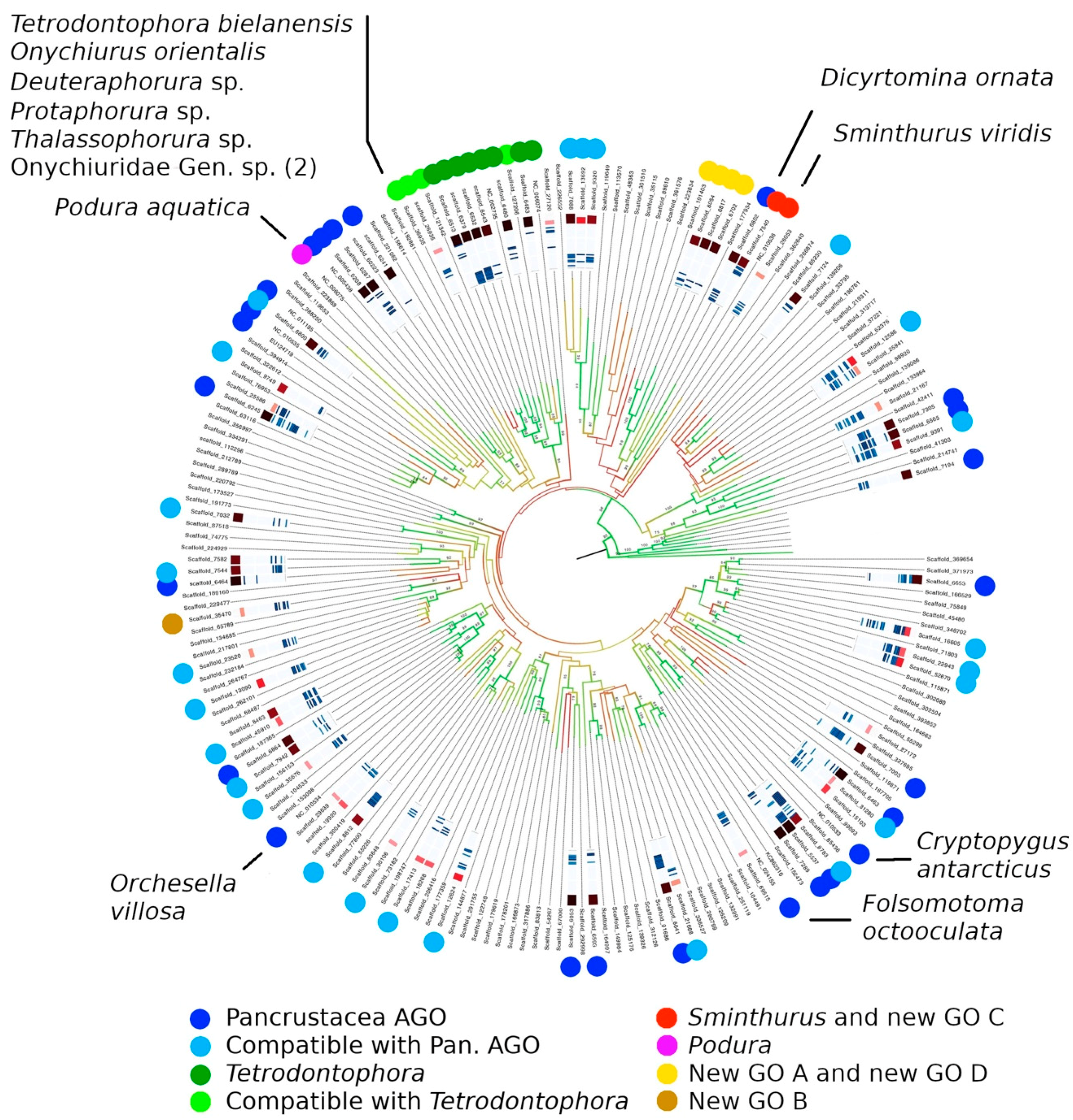
3.5. Gene Order
4. Discussion
4.1. Structure and Compositional Biases in the Two New Genomes
4.2. Phylogenetic Analysis
4.3. Dating Analysis
4.4. Gene Order
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Yoshii, R. Collembola of Himalaya. J. Coll. Arts Sci. Chiba Univ. 1966, 4, 461–531. [Google Scholar]
- Wise, K.A.J. Collembola (Springtails). In Entomology of Antarctica; Antarctic Research Series; American Geophysical Union (AGU): Washington, DC, USA, 1967; Volume 10, pp. 123–148. ISBN 978-1-118-66869-6. [Google Scholar]
- Hopkin, S.P. Biology of the Springtails: (Insecta: Collembola); OUP Oxford: Oxford, UK, 1997; pp. 1–330. ISBN 978-0-19-158925-6. [Google Scholar]
- Cicconardi, F.; Fanciulli, P.P.; Emerson, B.C. Collembola, the biological species concept and the underestimation of global species richness. Mol. Ecol. 2013, 22, 5382–5396. [Google Scholar] [CrossRef] [PubMed]
- Hirst, S.; Maulik, S. On some Arthropod Remains from the Rhynie Chert (Old Red Sandstone). Geol. Mag. 1926, 63, 69–71. [Google Scholar] [CrossRef]
- Whalley, P.; Jarzembowski, E.A. A new assessment of Rhyniella, the earliest known insect, from the Devonian of Rhynie, Scotland. Nature 1981, 291, 317. [Google Scholar] [CrossRef]
- Greenslade, P.; Whalley, P. The systematic position of Rhyniella praecursor hirst and maulik (Collembola), the earliest known hexapod. In Proceedings of the Second International Seminar on Apterygota, Siena, Italy, 4–6 September 1986; pp. 319–323. [Google Scholar]
- Greenslade, P.J.M. Reply to R. A. Crowson’s Comments on Insecta of the Rhynie Chert (1985 Entomol. Gener. 11 (1/2): 097–098). Entomol. Gen. 1988, 13, 115–117. [Google Scholar] [CrossRef]
- Nardi, F.; Spinsanti, G.; Boore, J.L.; Carapelli, A.; Dallai, R.; Frati, F. Hexapod origins: Monophyletic or Paraphyletic? Science 2003, 299, 1887–1889. [Google Scholar] [CrossRef] [PubMed]
- Carapelli, A.; Liò, P.; Nardi, F.; van der Wath, E.; Frati, F. Phylogenetic analysis of mitochondrial protein coding genes confirms the reciprocal paraphyly of Hexapoda and Crustacea. BMC Evol. Biol. 2007, 7, S8. [Google Scholar] [CrossRef]
- Borner, C. Das System der Collembolen, nebst beschreibungen neuer Collembolen des Hamburger Naturhistorichen Museums. Mitteilungen aus dem Naturhistorischen Museum in Hamburg 1906, 23, 147–188. [Google Scholar]
- Cassagnau, P. La Phylogenie des Collemboles à la lumiere des structures endocrines retrocerebrales. In Proceedings of the I Symposio International de Zoofilogenia. Facultad de Ciencias; Universidad de Salamanca: Salamanca, Spain, 1971; pp. 333–349. [Google Scholar]
- Massoud, Z. Essai de synthese sur la phylogenie des Collemboles. Rev. Ecol. Biol. Sol. 1976, 13, 241–252. [Google Scholar]
- D’Haese, C.A. Morphological appraisal of Collembola phylogeny with special emphasis on Poduromorpha and a test of the aquatic origin hypothesis. Zool. Scr. 2003, 32, 563–586. [Google Scholar] [CrossRef]
- Zhang, F.; Deharveng, L. First instar tibiotarsal chaetotaxy supports the Entomobryidae and Symphypleona (Collembola) forming a cluster in a phylogenetic tree. Zootaxa 2015, 3955, 487–504. [Google Scholar] [CrossRef] [PubMed]
- Carapelli, A.; Convey, P.; Nardi, F.; Frati, F. The mitochondrial genome of the antarctic springtail Folsomotoma octooculata (Hexapoda; Collembola), and an update on the phylogeny of collembolan lineages based on mitogenomic data. Entomologia 2014, 2, 46–55. [Google Scholar] [CrossRef]
- Luan, Y.-X.; Mallatt, J.M.; Xie, R.-D.; Yang, Y.-M.; Yin, W.-Y. The phylogenetic positions of three basal-hexapod groups (Protura, Diplura, and Collembola) based on ribosomal RNA gene sequences. Mol. Biol. Evol. 2005, 22, 1579–1592. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.; Cruaud, C.; D’Haese, C.A. Unexpected diversity in Neelipleona revealed by molecular phylogeny approach (Hexapoda, Collembola). Soil Org. 2011, 83, 383–398. [Google Scholar]
- Von Reumont, B.M.; Meusemann, K.; Szucsich, N.U.; Dell’Ampio, E.; Gowri-Shankar, V.; Bartel, D.; Simon, S.; Letsch, H.O.; Stocsits, R.R.; Luan, Y.; et al. Can comprehensive background knowledge be incorporated into substitution models to improve phylogenetic analyses? A case study on major arthropod relationships. BMC Evol. Biol. 2009, 9, 119. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Bu, Y.; Luan, Y.-X. Phylogenetic Relationships of Basal Hexapods Reconstructed from Nearly Complete 18S and 28S rRNA Gene Sequences. Zoolog. Sci. 2008, 25, 1139–1145. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Gao, Y.; Yin, W.; Luan, Y. Molecular phylogeny of Collembola inferred from ribosomal RNA genes. Mol. Phylogenet. Evol. 2008, 49, 728–735. [Google Scholar] [CrossRef] [PubMed]
- Boore, J.L. Animal mitochondrial genomes. Nucleic Acids Res. 1999, 27, 1767–1780. [Google Scholar] [CrossRef]
- Boore, J.L.; Lavrov, D.V.; Brown, W.M. Gene translocation links insects and crustaceans. Nature 1998, 392, 667. [Google Scholar] [CrossRef]
- Boore, J.L.; Brown, W.M. Big trees from little genomes: Mitochondrial gene order as a phylogenetic tool. Curr. Opin. Genet. Dev. 1998, 8, 668–674. [Google Scholar] [CrossRef]
- Nardi, F.; Carapelli, A.; Fanciulli, P.P.; Dallai, R.; Frati, F. The complete mitochondrial DNA sequence of the basal hexapod Tetrodontophora bielanensis: Evidence for heteroplasmy and tRNA translocations. Mol. Biol. Evol. 2001, 18, 1293–1304. [Google Scholar] [CrossRef] [PubMed]
- Cicconardi, F.; Borges, P.A.V.; Strasberg, D.; Oromí, P.; López, H.; Pérez Delgado, A.J.; Casquet, J.; Caujapé Castells, J.; Fernández Palacios, J.M.; Thébaud, C.; et al. MtDNA metagenomics reveals large-scale invasion of belowground arthropod communities by introduced species. Mol. Ecol. 2017, 26, 3104–3115. [Google Scholar] [CrossRef] [PubMed]
- Carapelli, A.; Fanciulli, P.P.; Frati, F.; Leo, C. Mitogenomic data to study the taxonomy of Antarctic springtail species (Hexapoda: Collembola) and their adaptation to extreme environments. Polar Biol. 2019, 42, 715–732. [Google Scholar] [CrossRef]
- Swofford, D.L. Phylogenetic Analysis Using Parsimony (*and Other Methods); Sinauer Associates: Sunderland, MA, USA, 2003. [Google Scholar]
- Hassanin, A.; Léger, N.; Deutsch, J. Evidence for Multiple Reversals of Asymmetric Mutational Constraints during the Evolution of the Mitochondrial Genome of Metazoa, and Consequences for Phylogenetic Inferences. Syst. Biol. 2005, 54, 277–298. [Google Scholar] [CrossRef] [PubMed]
- Bernt, M.; Donath, A.; Jühling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Pütz, J.; Middendorf, M.; Stadler, P.F. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Ratnasingham, S.; Hebert, P.D.N. BOLD: The Barcode of Life Data System. Mol. Ecol. Notes 2007, 7, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Wernersson, R.; Pedersen, A.G. RevTrans: Multiple alignment of coding DNA from aligned amino acid sequences. Nucleic Acids Res. 2003, 31, 3537–3539. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Castresana, J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 2000, 17, 540–552. [Google Scholar] [CrossRef] [PubMed]
- Lanfear, R.; Frandsen, P.B.; Wright, A.M.; Senfeld, T.; Calcott, B. PartitionFinder 2: New Methods for Selecting Partitioned Models of Evolution for Molecular and Morphological Phylogenetic Analyses. Mol. Biol. Evol. 2017, 34, 772–773. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [PubMed]
- Suchard, M.A.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018, 4, vey016. [Google Scholar] [CrossRef] [PubMed]
- Rehm, P.; Borner, J.; Meusemann, K.; von Reumont, B.M.; Simon, S.; Hadrys, H.; Misof, B.; Burmester, T. Dating the arthropod tree based on large-scale transcriptome data. Mol. Phylogenet. Evol. 2011, 61, 880–887. [Google Scholar] [CrossRef] [PubMed]
- Rota-Stabelli, O.; Daley, A.C.; Pisani, D. Molecular timetrees reveal a Cambrian colonization of land and a new scenario for ecdysozoan evolution. Curr. Biol. 2013, 23, 392–398. [Google Scholar] [CrossRef]
- Koch, M. Monophyly and phylogenetic position of the Diplura (Hexapoda). Pedobiologia 1997, 41, 9–12. [Google Scholar]
- Leo, C.; Nardi, F.; Frati, F.; Fanciulli, P.P.; Cucini, C.; Vitale, M.; Brunetti, C.; Carapelli, A. The mitogenome of the jumping bristletail Trigoniophthalmus alternatus (Insecta, Microcoryphia) and the phylogeny of insect early-divergent lineages. Mitochondrial DNA B. 2019, 4, 2855–2856. [Google Scholar] [CrossRef]
- Westoll, T.S. Northern Britain. In A correlation of the Devonian Rocks of the British Isles; Geological Society of London Special Report; Geological Society: London, UK, 1977; Volume 8, pp. 66–93. [Google Scholar]
- Garrouste, R.; Clément, G.; Nel, P.; Engel, M.S.; Grandcolas, P.; D’Haese, C.; Lagebro, L.; Denayer, J.; Gueriau, P.; Lafaite, P.; et al. A complete insect from the Late Devonian period. Nature 2012, 488, 82–85. [Google Scholar] [CrossRef]
- Rick, E.F. An entomobryid collembolan (Hexapoda: Collembola) from the Lower Permian of Southern Africa. Paleontol. Afr. 1976, 19, 141–143. [Google Scholar]
- Belica, M.E.; Tohver, E.; Poyatos-Moré, M.; Flint, S.; Parra-Avila, L.A.; Lanci, L.; Denyszyn, S.; Pisarevsky, S.A. Refining the chronostratigraphy of the Karoo Basin, South Africa: Magnetostratigraphic constraints support an early Permian age for the Ecca Group. Geophys. J. Int. 2017, 211, 1354–1374. [Google Scholar] [CrossRef]
- Lavrov, D.V. Key transitions in animal evolution: A mitochondrial DNA perspective. Integr. Comp. Biol. 2007, 47, 734–743. [Google Scholar] [CrossRef]
- Bradshaw, P.C.; Rathi, A.; Samuels, D.C. Mitochondrial-encoded membrane protein transcripts are pyrimidine-rich while soluble protein transcripts and ribosomal RNA are purine-rich. BMC Genomics 2005, 6, 136. [Google Scholar] [CrossRef] [PubMed]
- Henning, W. Insect Phylogeny; Wiley: New York, NY, USA, 1981; pp. 1–514. [Google Scholar]
- Yu, D.; Zhang, F.; Stevens, M.I.; Yan, Q.; Liu, M.; Hu, F. New insight into the systematics of Tomoceridae (Hexapoda, Collembola) by integrating molecular and morphological evidence. Zool. Scr. 2016, 45, 286–299. [Google Scholar] [CrossRef]
- Edwards, D.; Selden, P.A.; Richardson, J.B.; Axe, L. Coprolites as evidence for plant–animal interaction in Siluro–Devonian terrestrial ecosystems. Nature 1995, 377, 329–331. [Google Scholar] [CrossRef]
- Nardi, F.; Carapelli, A.; Boore, J.L.; Roderick, G.K.; Dallai, R.; Frati, F. Domestication of olive fly through a multi-regional host shift to cultivated olives: Comparative dating using complete mitochondrial genomes. Mol. Phylogenet. Evol. 2010, 57, 678–686. [Google Scholar] [CrossRef] [PubMed]
- Torricelli, G.; Carapelli, A.; Convey, P.; Nardi, F.; Boore, J.L.; Frati, F. High divergence across the whole mitochondrial genome in the pan-Antarctic springtail Friesea grisea: Evidence for cryptic species? Gene 2010, 449, 30–40. [Google Scholar] [CrossRef] [PubMed]
- Cicconardi, F.; Nardi, F.; Emerson, B.C.; Frati, F.; Fanciulli, P.P. Deep phylogeographic divisions and long-term persistence of forest invertebrates (Hexapoda: Collembola) in the North-Western Mediterranean basin. Mol. Ecol. 2010, 19, 386–400. [Google Scholar] [CrossRef]
- Von Saltzwedel, H.; Scheu, S.; Schaefer, I. Genetic structure and distribution of Parisotoma notabilis (Collembola) in Europe: Cryptic diversity, split of lineages and colonization patterns. PLoS ONE 2017, 12, e0170909. [Google Scholar] [CrossRef]
- Greenslade, P. An antarctic biogeographical anomaly resolved: The true identity of a widespread species of Collembola. Polar Biol. 2018, 41, 969–981. [Google Scholar] [CrossRef]
- Greenslade, P. A new species of Friesea (Collembola: Neanuridae) from the Antarctic Continent. J. Nat. Hist. 2018, 52, 2197–2207. [Google Scholar] [CrossRef]
- Tully, T.; D’Haese, C.A.; Richard, M.; Ferrière, R. Two major evolutionary lineages revealed by molecular phylogeny in the parthenogenetic collembola species Folsomia candida. Pedobiologia 2006, 50, 95–104. [Google Scholar] [CrossRef]
- Tillyard, R.J. Some remarks on the Devonian Fossil insects from the Rhynie Chert Beds, Old Red Sandstone. Trans. R. Entomol. Soc. Lond. 1928, 76, 65–71. [Google Scholar] [CrossRef]
- Massoud, Z. Contribution à l’étude de Rhyniella praecursor Hirst et Maulik. Rev. Ecol. Biol. Sol 1967, 4, 497–505. [Google Scholar]
- Scourfield, D.J. The oldest known fossil insect (Rhyniella praecursor Hirst & Maulik)—Further details from additional specimens. Proc. Linn. Soc. Lond. 1940, 152, 113–131. [Google Scholar]
- Scourfield, D.J. The oldest known fossil insect. Nature 1940, 145, 799–801. [Google Scholar] [CrossRef]
- Janssens, F. Checklist of the Collembola of the World. Available online: https://www.collembola.org/ (accessed on 17 July 2019).
- Cook, C.E.; Yue, Q.; Akam, M. Mitochondrial genomes suggest that hexapods and crustaceans are mutually paraphyletic. Proc. R. Soc. B Biol. Sci. 2005, 272, 1295–1304. [Google Scholar] [CrossRef] [PubMed]
- Crampton-Platt, A.; Yu, D.W.; Zhou, X.; Vogler, A.P. Mitochondrial metagenomics: Letting the genes out the bottle. GigaScience 2016, 5, 15. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Accession Number/Scaffold | Family (Class for Outgroup) | Species | PCGs | Genes | GO |
|---|---|---|---|---|---|
| NC_039558.1 | Bourletiellidae | Bourletiella arvalis | 13 | 37 | GO1 |
| MG701393 | Dicyrtomidae | Dicyrtomina saundersi | 13 | 37 | GO1 |
| s6802 | Dicyrtomidae | gen. sp. 1 | 13 | 37 | GO1 |
| NC_010534.1 | Entomobryidae | Orchesella villosa | 13 | 37 | GO1 |
| NC_032283 | Entomobryidae | Orchesella cincta | 13 | 37 | GO1 |
| s6241 | Hypogastruridae | gen. sp. | 13 | 37 | GO1 |
| NC_005438 | Hypogastrurudae | Gomphiocephalus hodgsoni | 13 | 37 | GO1 |
| NC_010533.1 | Isotomidae | Cryptopygus antarcticus | 13 | 37 3 | GO1 |
| NC_024155.1 | Isotomidae | Folsomotoma octooculata | 13 | 37 | GO1 |
| KU198392 | Isotomidae | Folsomia candida | 13 | 37 | GO1 |
| NC_037610.1 | Isotomidae | Cryptopygus terranovus | 13 | 37 | GO1 |
| s6653 | Isotomidae | Folsomia candida1 | 13 | 37 | GO1 |
| s8783 | Isotomidae | Parisotoma notabilis L1 | 12 | 32 | GO1 |
| s5537 | Isotomidae | Parisotoma notabilis L2 | 13 | 37 | GO1 |
| s7289 | Isotomidae | Desoria trispinata1 | 13 | 37 | GO1 |
| s6464 | Entomobryidae | Lepidocyrtus curvicollis | 13 | 37 | GO1 |
| NC_010535.1 | Neanuridae | Friesea antarctica AP 2 | 13 | 37 | GO1 |
| EU124719.1 | Neanuridae | Friesea antarctica VL 2 | 13 | 37 | GO1 |
| NC_011195.1 | Neanuridae | Bilobella aurantiaca | 13 | 37 4 | GO1 |
| MH155200 | Neelidae | Neelus murinus | 13 | 34 | GO1 |
| s6565 | Neelidae | Megalothorax minimus | 13 | 37 5 | GO1 |
| s7305 | Neelidae | gen. sp. | 13 | 37 | GO1 |
| NC_002735.1 | Onychiuridae | Tetrodontophora bielanensis | 13 | 37 | GO2 |
| NC_006074.1 | Onychiuridae | Onychiurus orientalis | 13 | 34 | GO2 |
| s6480 | Onychiuridae | Deuteraphorura sp. | 13 | 37 | GO2 |
| s6543 | Onychiuridae | Thalassophorura sp. 1 | 13 | 37 | GO2 |
| s6532 | Onychiuridae | gen. sp. | 13 | 37 | GO2 |
| s6379 | Onychiuridae | gen. sp. | 13 | 37 | GO2 |
| NC_006075.1 | Poduridae | Podura aquatica | 13 | 34 | GO4 |
| NC_010536.1 | Sminthuridae | Sminthurus viridis | 13 | 37 | GO3 |
| s7124 | Tullbergiidae | gen. sp. | 13 | 35 5 | GO1 |
| NC_000844 | Branchiopoda | Daphnia pulex | 13 | - | - |
| NC_007214 | Diplura | Japyx solifugus | 13 | - | - |
| NC_010532 | Microcoryphia | Trigoniophthalmus alternatus | 13 | - | - |
| Priors 1 | Collembola | Poduromorpha | Entomobryiomorpha | Symphypleona | Neelipleona | Diversification of 4 Groups 2 | Phylogeny 4 |
|---|---|---|---|---|---|---|---|
| 1: N510 2: N485 | 391 (368–413) | 356 (332–381) | 295 (270–323) | 248 (222–275) | 335 (295–367) | 348–391 (323–413) | ((S,E),P),N) |
| 1: N510 2: N485 | 382 (356–406) | 349 (323–375) | 265 (237–292) | 227 (201–254) | 334 (305–363) | 328–382 (301–406) | ((S,E),P),N) |
| 1: N510 2: N485 5: G280 4: G391 | 437 (426–448) | 387 3 (368–404) | 392 (391–396) | 296 (270–322) | 352 (326–376) | 419–437 3 (432–448) | ((S,E),(P,N)) |
| 1: N510 2: N485 5: G280 4: G391 | 437 (426–452) | 414 3 (395–433) | 392 (391–396) | 283 (250–315) | 367 (336–400) | 421–437 3 (411–452) | ((S,E),(P,N)) |
| 1: N510 2: N485 5: G280 | 404 (386–423) | 358 3 (338–377) | 329 (314–346) | 266 (242–292) | 335 (304–375) | 374–404 3 (354–423) | ((S,E),(P,N)) |
| 1: N510 2: N485 5: G280 | 409 (390–427) | 379 (359–399) | 322 (309–337) | 252 (224–279) | 357 (330–384) | 366–409 (348–427) | ((S,E),P),N) |
| 1: N510 2: N485 5: G280 3: G391 | 414 (403–426) | 369 3 (352–386) | 339 (324–355) | 278 (255–301) | 336 (310–361) | 394–414 3 (391–426) | ((S,E),(P,N)) |
| 1: N510 2: N485 5: G280 3: G391 | 425 (409–439) | 398 (382–414) | 333 (317–349) | 269 (240–296) | 369 (337–396) | 393–425 (391–439) | ((S,E),P),N) |
| 1: N510 2: N485 4: G280 | 391 (340–435) | 357 (316–408) | 294 (280–350) | 248 (202–298) | 337 (258–392) | 348–391 (310–435) | ((S,E),P),N) |
| 1: N510 2: N485 4: G280 | 391 (371–411) | 360 (338–380) | 280 (280–298) | 236 (212–262) | 342 (314–368) | 341–391 (322–411) | ((S,E),P),N) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leo, C.; Carapelli, A.; Cicconardi, F.; Frati, F.; Nardi, F. Mitochondrial Genome Diversity in Collembola: Phylogeny, Dating and Gene Order. Diversity 2019, 11, 169. https://doi.org/10.3390/d11090169
Leo C, Carapelli A, Cicconardi F, Frati F, Nardi F. Mitochondrial Genome Diversity in Collembola: Phylogeny, Dating and Gene Order. Diversity. 2019; 11(9):169. https://doi.org/10.3390/d11090169
Chicago/Turabian StyleLeo, Chiara, Antonio Carapelli, Francesco Cicconardi, Francesco Frati, and Francesco Nardi. 2019. "Mitochondrial Genome Diversity in Collembola: Phylogeny, Dating and Gene Order" Diversity 11, no. 9: 169. https://doi.org/10.3390/d11090169
APA StyleLeo, C., Carapelli, A., Cicconardi, F., Frati, F., & Nardi, F. (2019). Mitochondrial Genome Diversity in Collembola: Phylogeny, Dating and Gene Order. Diversity, 11(9), 169. https://doi.org/10.3390/d11090169