New Insights Into Nematode DNA-metabarcoding as Revealed by the Characterization of Artificial and Spiked Nematode Communities


Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Preparation
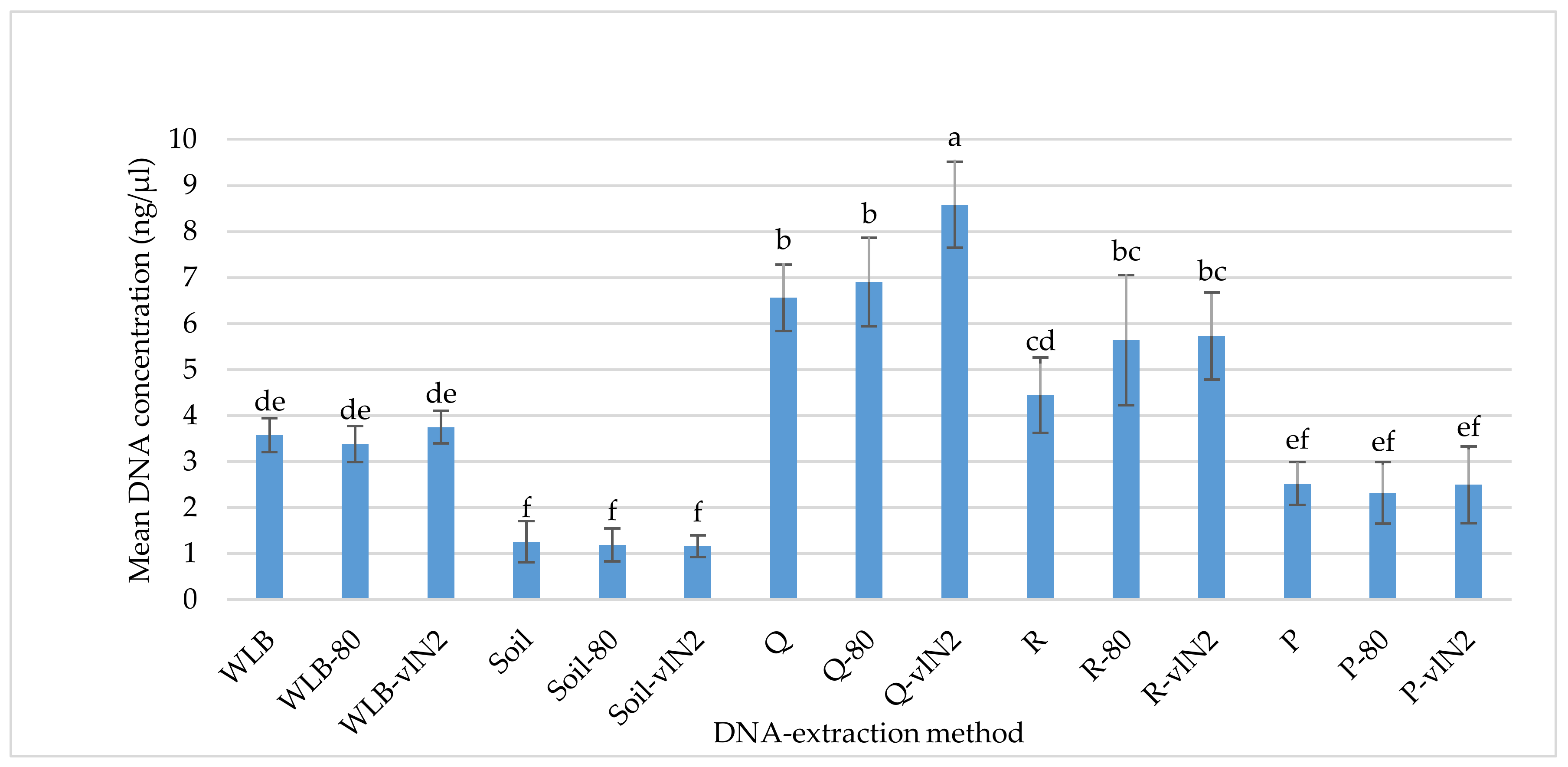
2.2. DNA-Extraction Test
2.3. Nematode Reference Sequence Database
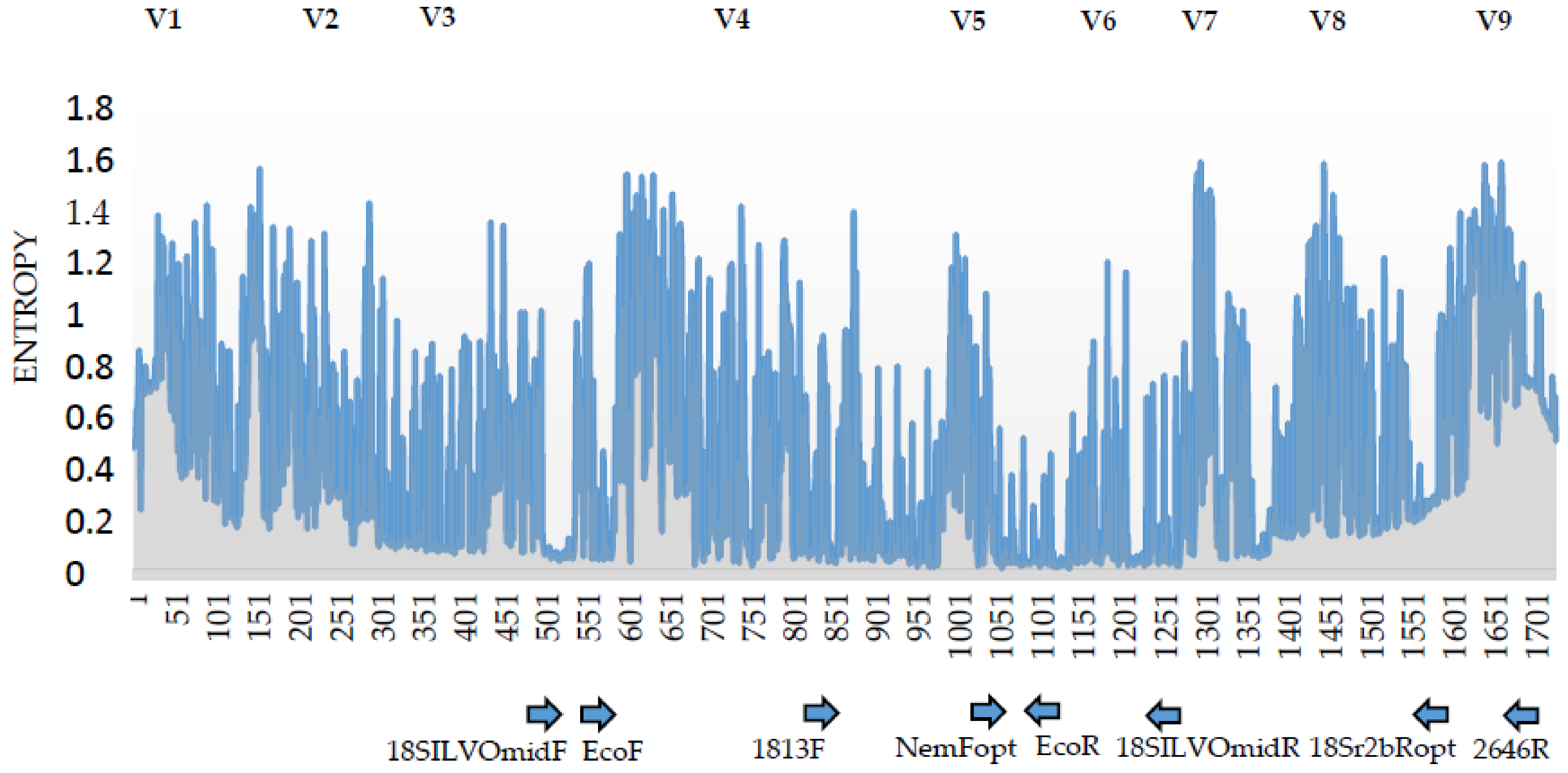
2.4. 18S rDNA Primer Pairs for a Amplicon Sequencing
2.5. NGS Library Preparation
2.6. Data Analysis
3. Results
3.1. DNA-extraction Test
3.2. Analysis of the 18S rDNA Primer Pairs for Nematode DNA-Metabarcoding
3.3. DNA-Metabarcoding Data
3.3.1. The Number of Reads Per Sample and Taxonomic Assignment Depends on the DNA-Extraction Method and Primer Pair
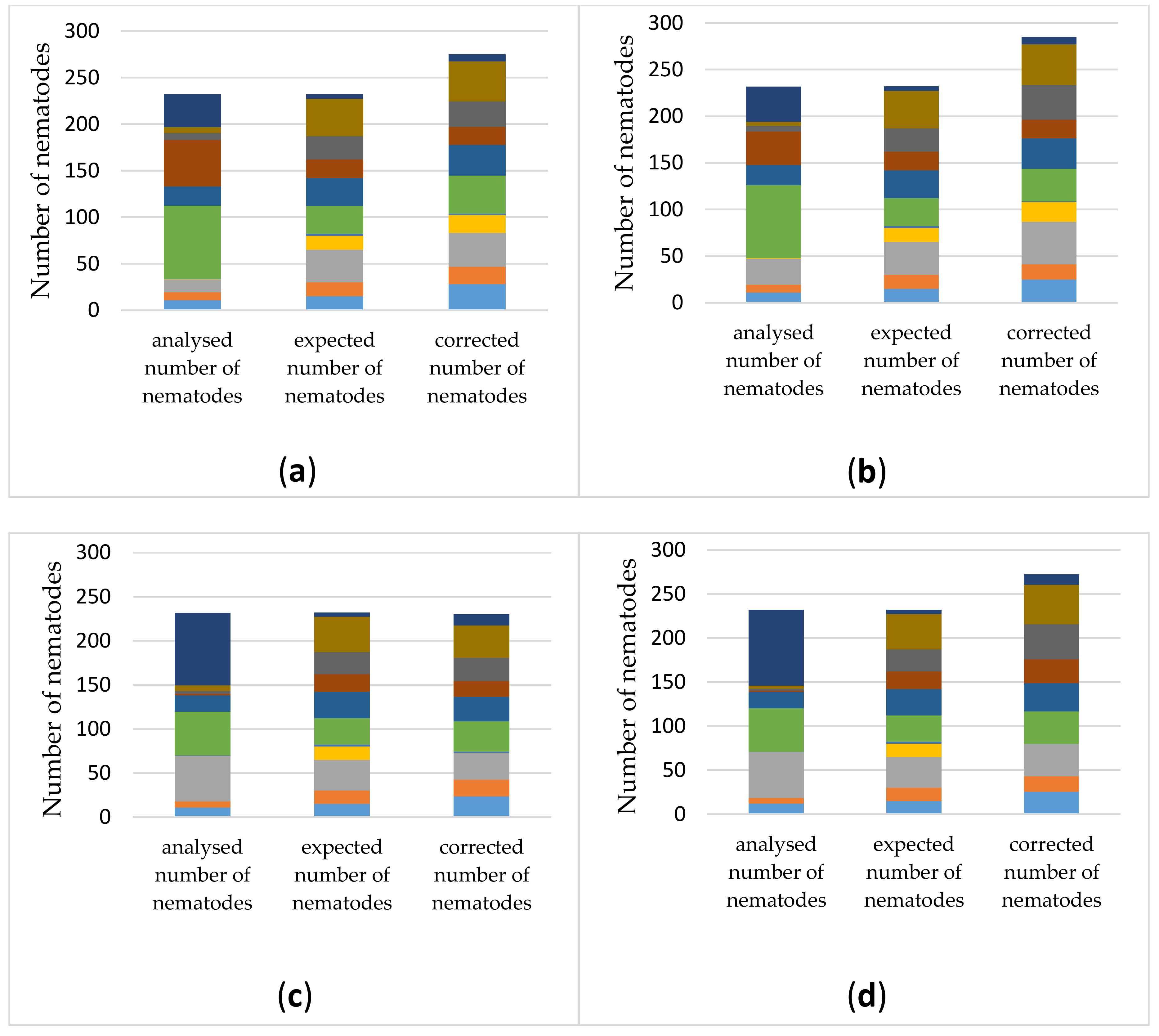
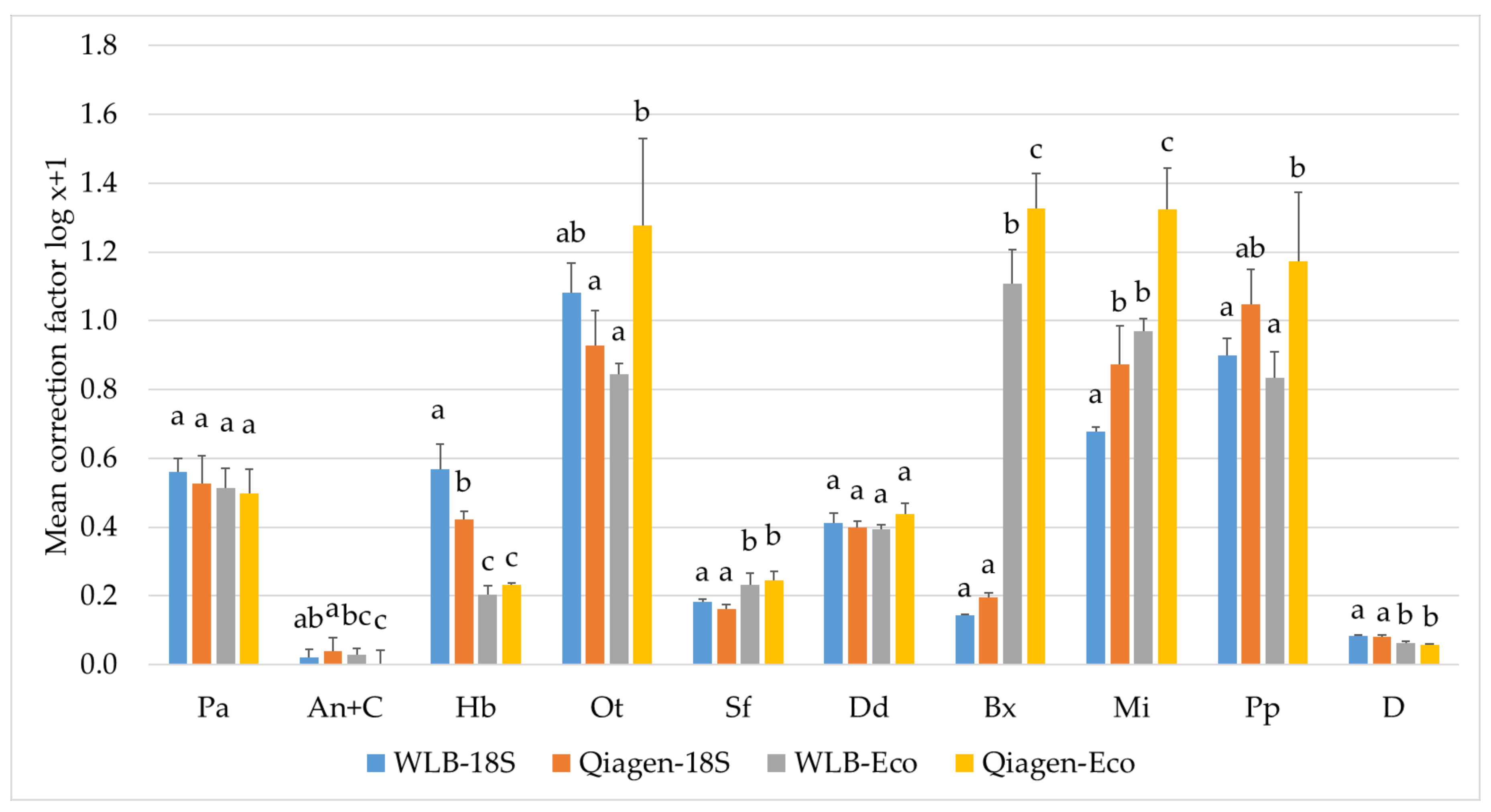
3.3.2. Estimating Relative Abundances Is More Accurate When Using Protocol-Specific Correction Factors
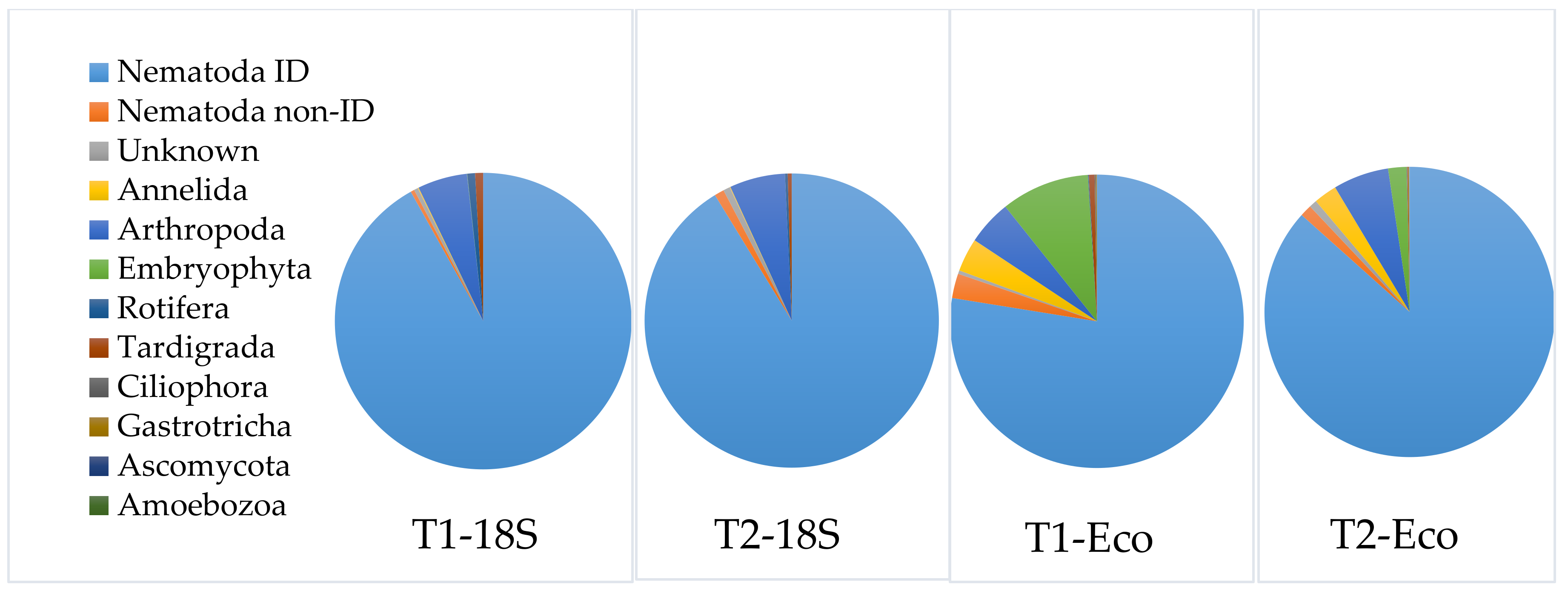
3.3.3. Application of the Selected DNA-Extraction Methods, Primer Sets and Correction Factors on a Spiked Biological Sample
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Yeates, G.W. Soil nematodes in terrestrial ecosystems. J. Nematol. 1979, 11, 213–229. [Google Scholar] [PubMed]
- Bongers, T. The maturity index: An ecological measure of environmental disturbance based on nematode species composition. Oecologia 1990, 83, 14–19. [Google Scholar] [CrossRef]
- Ferris, H.; Bongers, T.; de Goede, R.G.M. A framework for soil food web diagnostics: Extension of the nematode faunal analysis concept. Appl. Soil Ecol. 2001, 18, 13–29. [Google Scholar] [CrossRef]
- Nannipieri, P.; Grego, S.; Ceccanti, B. Ecological significance of the biological activity in soil. In Soil Biochemistry; Bollag, J.-M., Stotzky, G., Eds.; Marcel Dekker Inc.: New York, NY, USA, 1990; Volume 6, pp. 293–355. [Google Scholar]
- Ritz, K.; Black, H.I.J.; Campbell, C.D.; Harris, J.A.; Wood, C. Selecting biological indicators for monitoring soils: A framework for balancing scientific and technical opinion to assist policy development. Ecol. Ind. 2009, 9, 1212–1221. [Google Scholar] [CrossRef]
- Griffiths, B.S.; Römbke, J.; Schmelz, R.M.; Scheffczyk, A.; Faber, J.H.; Bloem, J.; Pérès, G.; Cluzeau, D.; Chabbi, A.; Suhadolc, M.; et al. Selecting cost effective and policy-relevant biological indicators for European monitoring of soil biodiversity and ecosystem function. Ecol. Indic. 2016, 69, 213–223. [Google Scholar] [CrossRef]
- Jones, J.T.; Haegeman, A.; Danchin, E.G.J.; Gaur, H.S.; Helder, J.; Jones, M.G.K.; Kikuchi, T.; Manzanilla-López, R.; Palomares-Rius, J.E.; Wesemael, W.M.L.; et al. Top 10 plant-parasitic nematodes in molecular plant pathology. Mol. Plant Pathol. 2013, 14, 946–961. [Google Scholar] [CrossRef] [PubMed]
- Ritz, K.; Trudgill, D.L. Utility of nematode community analysis as an integrated measure of the functional state of soils: Perspectives and challenges. Plant Soil 1999, 212, 1–11. [Google Scholar] [CrossRef]
- Neher, D. Role of nematodes in soil health and their use as indicators. J. Nematol. 2001, 33, 161–168. [Google Scholar]
- Porazinska, D.L.; Giblin-Davis, R.M.; Faller, L.; Farmerie, W.; Kanzaki, N.; Morris, K.; Powers, T.O.; Tucker, A.E.; Sung, W.; Thomas, W.K. Evaluating high-throughput sequencing as a method for metagenomics analysis of nematode diversity. Mol. Ecol. Resour. 2009, 9, 1439–1450. [Google Scholar] [CrossRef]
- Porazinska, D.L.; Giblin-Davis, R.M.; Esquivel, A.; Powers, T.O.; Sung, W.; Thomas, W.K. Ecometagenetics confirms high tropical rainforest nematode diversity. Mol. Ecol. 2010, 19, 5521–5530. [Google Scholar] [CrossRef]
- Porazinska, D.L.; Giblin-Davis, R.M.; Sung, W.; Thomas, W.K. Linking operational clustered taxonomic units (OCTUs) from parallel ultra sequencing (PUS) to nematode species. Zootaxa 2010, 2427, 55–63. [Google Scholar] [CrossRef]
- Darby, B.J.; Todd, T.C.; Herman, M.A. High-throughput amplicon sequencing of rRNA genes requires a copy number correction to accurately reflect the effects of management practices on soil nematode community structure. Mol. Ecol. 2013, 22, 5456–5471. [Google Scholar] [CrossRef]
- Sapkota, R.; Nicolaisen, M. High-throughput sequencing of nematode communities from total soil DNA-extractions. BMC Ecol. 2015, 15, 3. [Google Scholar] [CrossRef] [PubMed]
- Peham, T.; Steiner, F.M.; Schlick-Steiner, B.C.; Arthofer, W. Are we ready to detect nematode diversity by next generation sequencing? Ecol. Evol. 2016, 7, 4147–4151. [Google Scholar] [CrossRef]
- Griffiths, B.S.; de Groot, G.A.; Laros, I.; Stone, D.; Geisen, S. The need for standardisation: Exemplified by a description of the diversity, community structure and ecological indices of soil nematodes. Ecol. Indic. 2018, 87, 43–46. [Google Scholar] [CrossRef]
- Wu, T.; Ayres, E.; Li, G.; Bardgett, R.D.; Wall, D.H.; Garey, J.R. Molecular profiling of soil animal diversity in natural ecosystems: Incongruence of molecular and morphological results. Soil Biol. Biochem. 2009, 41, 849–857. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High resolution sample inference from illumine amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- O’Bannon, J.H.; Taylor, A.L. Migratory endoparasitic nematodes reared on carrot disks. Phytopathology 1968, 58, 385. [Google Scholar]
- Kobayashi, T.; Saski, K.; Mamiya, Y. Fungi associated with Bursaphelenchus lignicolus the pine wood nematode. J. Jpn. For. Soc. 1975, 57, 184–193. [Google Scholar] [CrossRef]
- Brenner, S. The genetics of Caenorhabditis elegans. Genetics 1974, 77, 71–94. [Google Scholar] [CrossRef]
- Decraemer, W.; Brown, D.; Karanastasi, E.; Zheng, J.; De Almeida, M.T.M. Interspecific differences in the fine structure of the body cuticle of Trichodoridae Thorne, 1935 (Nematoda: Diphtherophorina) and review of anchoring structures of the epidermis. Nematology 2001, 3, 525–533. [Google Scholar] [CrossRef]
- Treonis, A.M.; Unangst, S.K.; Kepler, R.M.; Buyer, J.S.; Cavigelli, M.A.; Mirsky, S.B.; Maul, J.E. Characterization of soil nematode communities in three cropping systems through morphological and DNA metabarcoding approaches. Sci. Rep. 2018, 8, 2004. [Google Scholar] [CrossRef]
- Hendrickx, G. An automatic apparatus for extracting free-living nematode stages from soil. Nematologica 1995, 41, 308. [Google Scholar]
- Holterman, M.; van der Wurff, A.; van den Elsen, S.; van Megen, H.; Bongers, T.; Holovachov, O.; Bakker, J.; Helder, J. Phylum-Wide Analysis of SSU rDNA Reveals Deep Phylogenetic Relationships among Nematodes and Accelerated Evolution towards Crown Clades. Mol. Biol. Evol. 2006, 23, 1792–1800. [Google Scholar] [CrossRef] [PubMed]
- Campos-Herrera, R.; El-Borai, F.E.; Stuart, R.J.; Graham, J.H.; Duncan, L.W. Entomopathogenic nematodes, phoretic Paenibacillus spp., and the use of real time quantitative PCR to explore soil food webs in Florida citrus groves. J. Invertebr. Pathol. 2011, 108, 30–39. [Google Scholar] [CrossRef] [PubMed]
- BioNumerics. Available online: http://www.applied-maths.com/ (accessed on 14 March 2019).
- Welcome to NVBI. Available online: https://www.ncbi.nlm.nih.gov/ (accessed on 14 March 2019).
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- Walters, W.A.; Caporaso, J.G.; Lauber, C.L.; Berg-Lyons, D.; Fierer, N.; Knight, R. PrimerProspector: De novo design and taxonomic analysis of PCR primers. Bioinformatics 2011, 27, 1159–1161. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Riaz, T.; Shehzad, W.; Viari, A.; Pompanon, F.; Taberlet, P.; Coissac, E. ecoPrimers: Inference of new DNA barcode markers from whole genome sequence analysis. Nucleic Acids Res. 2011, 39, e145. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna, Austria. Available online: https://www.r-project.org/ (accessed on 31 August 2017).
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naïve Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microb. 2007, 73, 5261–5267. [Google Scholar] [CrossRef] [PubMed]
- Neefs, J.M.; Van de Peer, Y.; De Rijk, P.; Chapelle, S.; De Wachter, R. Compilation of small ribosomal subunit structures. Nucleic Acids Res. 1993, 21, 3025–3049. [Google Scholar] [CrossRef] [PubMed]
- Thomsen, P.F.; Willerslev, E. Environmental DNA—An emerging tool in conservation for monitoring past and present biodiversity. Biol. Conserv. 2015, 183, 4–18. [Google Scholar] [CrossRef]
- Deiner, K.; Bik, H.M.; Mächler, E.; Seymour, M.; Lacoursière-Roussel, A.; Altermatt, F.; Creer, S.; Bista, I.; Lodge, D.M.; de Vere, N.; et al. Environmental DNA metabarcoding: Transforming how we survey animal and plant communities. Mol. Ecol. 2017, 26, 5872–5895. [Google Scholar] [CrossRef]
- Deiner, K.; Fronhofer, E.A.; Mächler, E.; Walser, J.-C.; Altermatt, F. Environmental DNA reveals that rivers are conveyer belts of biodiversity information. Nat. Commun. 2016, 7, 12544. [Google Scholar] [CrossRef]
- Carini, P.; Marsden, P.J.; Leff, J.W.; Morgan, E.E.; Strickland, M.S.; Fierer, N. Relic DNA is abundant in soil and obscures estimates of soil microbial diversity. Nat. Microbiol. 2016, 2, 16242. [Google Scholar] [CrossRef]
- Yeates, G.W.; Bongers, T.; de Goede, R.G.M.; Freckman, D.W.; Georgieva, S.S. Feeding habits in soil nematode families and genera-an outline for soil ecologists. J. Nematol. 1993, 3, 315–331. [Google Scholar]
- Donn, S.; Griffiths, B.S.; Neilson, R.; Daniell, T.J. DNA extraction from soil nematodes for multi-sample community studies. Appl. Soil Ecol. 2008, 38, 20–26. [Google Scholar] [CrossRef]
- Bird, A.F. The exoskeleton. In The Structure of Nematodes, 1st ed.; Academic Press: New York, NY, USA, 1971; pp. 45–79. [Google Scholar]
- Cordero, M.A.; Robbins, R.T.; Szalanski, A.L. Taxonomic and molecular identification of Mesocriconema and Criconemoides species (Nematoda: Criconematidae). J. Nematol. 2012, 44, 399–426. [Google Scholar]
- Macheriotou, L.; Guilini, K.; Bezerra, T.N.; Tytgat, B.; Nguyen, D.T.; Nguyen, T.X.P.; Noppe, F.; Armenteros, M.; Boufahja, F.; Rigaux, A.; et al. Metabarcoding free-living marine nematodes using curated 18S and CO1 reference sequence databases for species-level taxonomic assignments. Ecol. Evol. 2018, 9, 1211–1226. [Google Scholar] [CrossRef]
- Hadziavdic, K.; Lekang, K.; Lanzen, A.; Jonassen, I.; Thompson, E.M.; Troedsson, C. Characterization of the 18S rRNA gene for designing universal Eukaryote specific primers. PLoS ONE 2014, 9, e87624. [Google Scholar] [CrossRef] [PubMed]
- Van Megen, H.; Van den Elsen, S.; Holterman, M.; Karssen, G.; Mooyman, P.; Bongers, T.; Holovachov, O.; Bakker, J.; Helder, J. A phylogenetic tree of nematodes based on about 1200 full-length small subunit ribosomal DNA sequences. Nematology 2009, 11, 927–950. [Google Scholar] [CrossRef]
- Donn, S.; Neilson, R.; Griffiths, B.S.; Daniell, T.J. Greater coverage of the phylum Nematoda in SSU rDNA studies. Biol. Fertil. Soils 2011, 47, 333–339. [Google Scholar] [CrossRef]
- Stadhouders, R.; Pas, S.D.; Anber, J.; Voermans, J.; Mes, T.H.M.; Schutten, M. The effect of primer-template mismatches on the detection and quantification of nucleic acids using the 5′ nuclease assay. J. Mol. Diagn. 2010, 12, 109–117. [Google Scholar] [CrossRef]
- Smythe, A.B.; Nadler, S.A. Molecular phylogeny of Acrobeloides and Cephalobus (Nematoda: Cephalobidae) reveals paraphyletic taxa and recurrent evolution of simple labial morphology. Nematology 2006, 8, 819–836. [Google Scholar] [CrossRef]
- Piñol, J.; Mir, G.; Gomez-Polo, P.; Agusti, N. Universal and blocking primer mismatches limit the use of high-throughput DNA sequencing for the quantitative metabarcoding of arthropods. Mol. Ecol. Resour. 2015, 15, 819–830. [Google Scholar] [CrossRef]
- Page, A.P.; Johnstone, I.L. The cuticle. WormBook Online Rev. C. Elegans Boil. 2007, 1–15. [Google Scholar] [CrossRef]
- Bik, H.M.; Fournier, D.; Sung, W.; Bergeron, R.D.; Thomas, W.K. Intra-genomic variation in the ribosomal repeats of nematodes. PLoS ONE 2013, 8, e78230. [Google Scholar] [CrossRef] [PubMed]
- Blouin, M.S.; Yowell, C.A.; Courtney, C.H.; Dame, J.B. Substitution bias, rapid saturation, and the use of mtDNA for nematode systematics. Mol. Biol. Evol. 1998, 15, 1719–1727. [Google Scholar] [CrossRef] [PubMed]
- Deagle, B.E.; Jarman, S.N.; Coissac, E.; Pompanon, F.; Taberlet, P. DNA metabarcoding and the cytochrome c oxidase subunit I marker: Not a perfect match. Biol. Lett. 2014, 10, 20140562. [Google Scholar] [CrossRef] [PubMed]
- Lopes, E.A.; Roberts, D.M.; Blok, V.C. Variable ITS-copy number at different developmental stages of Meloidogyne hapla and M. chitwoodi. Eur. J. Plant Pathol. 2019. [Google Scholar] [CrossRef]
- Yang, C.; Wang, X.; Miller, J.A.; de Blécourt, M.; Ji, Y.; Yang, C.; Harrison, R.D.; Yu, D.W. Using metabarcoding to ask if easily collected soil and leaf-litter samples can be used as a general biodiversity indicator. Ecol. Indic. 2014, 46, 379–389. [Google Scholar] [CrossRef]
- Hajibabaei, M.; Shokralla, S.; Zhou, X.; Singer, G.A.C.; Baird, D.J. Environmental barcoding: A next-generation sequencing approach for biomonitoring applications using river benthos. PLoS ONE 2011, 6, e17497. [Google Scholar] [CrossRef] [PubMed]
- Emerson, B.; Cicconardi, F.; Fanciulli, P.P.; Shaw, P.J.A. Phylogeny, phylogeography, phylobetadiversity and the molecular analysis of biological communities. Philos. Trans. R. Soc. B 2011, 366, 2391–2402. [Google Scholar] [CrossRef] [PubMed]
- Bik, H.M.; Sung, W.; De Ley, P.; Baldwin, J.G.; Sharma, J.; Rocha-Olivares, A.; Thomas, W.K. Metagenetic community analysis of microbial eukaryotes illuminates biogeographic patterns in deep-sea and shallow water sediments. Mol. Ecol. 2012, 21, 1048–1059. [Google Scholar] [CrossRef]
- Purdy, K.J.; Hurd, P.J.; Moya-Larano, J.; Trimmer, M.; Oakley, B.B.; Woodward, G. Systems biology for ecology: From molecules to ecosystems. Adv. Ecol. Res. 2010, 43, 87–149. [Google Scholar] [CrossRef]
- Lamb, P.D.; Hunter, E.; Pinnegar, J.K.; Creer, S.; Davies, R.G.; Taylor, M.I. How quantitative is metabarcoding: A meta-analytical approach. Mol. Ecol. 2018. [Google Scholar] [CrossRef]
- Creer, S.; Deiner, K.; Frey, S.; Porazinska, D.; Taberlet, P.; Thomas, W.K.; Potter, C.; Bik, H.M. The ecologist’s field guide to sequence-based identification of biodiversity. Methods Ecol. Evol. 2016, 7, 1008–1018. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nematode Isolate and Genbank Accession Number | Taxonomic Lineage: Class/Order/Family | Code of Single-Species Sample | Number of Individuals in Single-Species Samples | Number of Individuals in Mock Community M20 | Number of Individuals in Mock Community Mvar | Number of Spiked Individuals in Nematode Suspension T1 | Number of Spiked Individuals in Nematode Suspension T2 |
|---|---|---|---|---|---|---|---|
| Acrobeloides nanus MH983014 | Chromadorea/ Rhabditida/ Cephalobidae | An | - | 20 | 10 | - | - |
| Bursaphelenchus xylophilus MH983015 | Chromadorea/ Tylenchida/ Aphelenchoididae | Bx | 30 | 20 | 20 | 40 | 1 |
| Cephalobus sp. MH983016 | Chromadorea/ Rhabditida/ Cephalobidae | C | - | 20 | 5 | - | - |
| Ditylenchus dipsaci MH983018 | Chromadorea/ Tylenchida/ Anguinidae | Dd | 30 | 20 | 30 | - | - |
| Heterorhabditis bacteriophora MH983019 | Chromadorea/ Rhabditida/ Heterorhabditidae | Hb | - | 20 | 35 | 5 | 40 |
| Meloidogyne incognita MH983020 | Chromadorea/ Tylenchida/ Meloidogynidae | Mi | - | 20 | 25 | - | - |
| Mesocriconema sp. MH983017 | Chromadorea/ Tylenchida/ Criconematidae | Cr | 10 | - | - | - | - |
| Mesorhabditis belari MH983024 | Chromadorea/ Rhabditida/ Rhabditidae | Rb | - | 20 | 15 | - | - |
| Oscheius tipulae MH983026 | Chromadorea/ Rhabditida/ Rhabditidae | Ot | - | 20 | 2 | - | - |
| Paratylenchus sp. MH983022 | Chromadorea/ Tylenchida/ Paratylenchidae | Par | 7 | - | - | - | - |
| Plectus acuminatus MH983021 | Chromadorea/ Plectida/ Plectidae | Pa | 30 | 20 | 15 | 10 | 10 |
| Pratylenchus penetrans MH983023 | Chromadorea/ Tylenchida/ Pratylenchidae | Pp | - | 20 | 40 | 20 | 20 |
| Steinernema feltiae MH983025 | Chromadorea/ Rhabditida/ Steinernematidae | Sf | - | 20 | 30 | - | - |
| Dorylaimidae fam. (not sequenced) | Enoplea/ Dorylaimida | D | - | 20 | 5 | - | - |
| Primer Code | Colloquial Primer Set Name | Nucleotide Sequence (5’-3’) | Amplicon Length (bp) | Application | Source |
|---|---|---|---|---|---|
| 18SILVOmidF | - | CAAGTCTGGTGCCAGCAG- | 763 1 | Sanger-sequencing | ILVO |
| 18SILVOmidR | GAGTCTCGCTCGTTATCGG | ||||
| 1813F | - | CTGCGTGAGAGGTGAAAT | 870 1 | Sanger-sequencing | [23] |
| 2646R | GCTACCTTGTTACGACTTTT | ||||
| NemFopt | Adapted primer set | GGGGWAGTATGGTTGCAAA | 489.9 2 | DNA-metabarcoding | [13] |
| 18Sr2bRopt | TGTGTACAAAKGRCAGGGAC | ||||
| EcoF | Eco primer set | GGTTAAAAMGYTCGTAGTTG | 510.4 2 | DNA-metabarcoding | This study |
| EcoR | TGGTGGTGCCCTTCCGTCA |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Waeyenberge, L.; de Sutter, N.; Viaene, N.; Haegeman, A. New Insights Into Nematode DNA-metabarcoding as Revealed by the Characterization of Artificial and Spiked Nematode Communities. Diversity 2019, 11, 52. https://doi.org/10.3390/d11040052
Waeyenberge L, de Sutter N, Viaene N, Haegeman A. New Insights Into Nematode DNA-metabarcoding as Revealed by the Characterization of Artificial and Spiked Nematode Communities. Diversity. 2019; 11(4):52. https://doi.org/10.3390/d11040052
Chicago/Turabian StyleWaeyenberge, Lieven, Nancy de Sutter, Nicole Viaene, and Annelies Haegeman. 2019. "New Insights Into Nematode DNA-metabarcoding as Revealed by the Characterization of Artificial and Spiked Nematode Communities" Diversity 11, no. 4: 52. https://doi.org/10.3390/d11040052
APA StyleWaeyenberge, L., de Sutter, N., Viaene, N., & Haegeman, A. (2019). New Insights Into Nematode DNA-metabarcoding as Revealed by the Characterization of Artificial and Spiked Nematode Communities. Diversity, 11(4), 52. https://doi.org/10.3390/d11040052