Taking Advantage of the Genomics Revolution for Monitoring and Conservation of Chondrichthyan Populations

{kind=link}
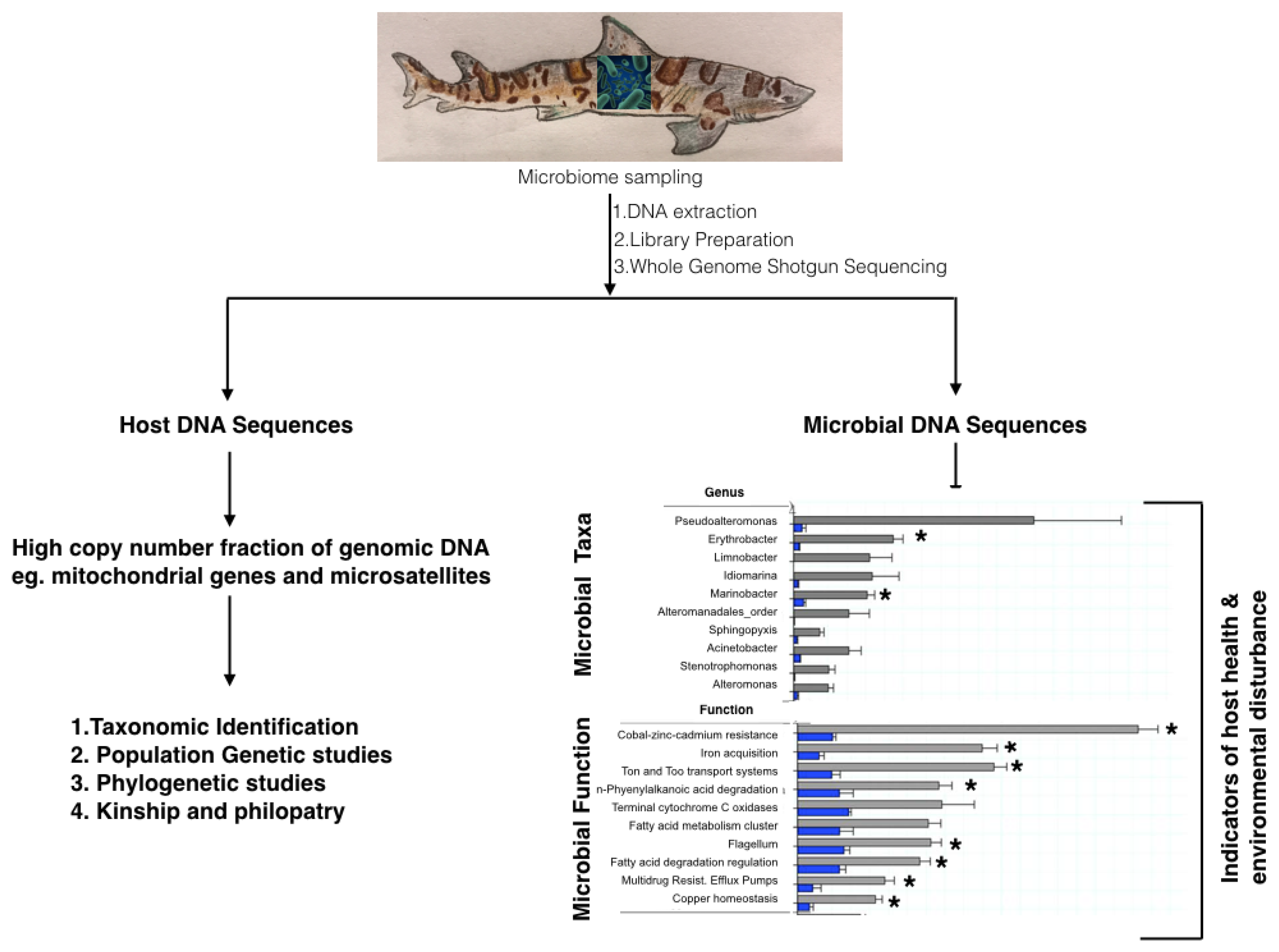{kind=link}
Abstract
1. Introduction
2. Genetics and Genomics for Chondrichthyan Biodiversity Assessments
2.1. Taxonomy, Phylogenetics and Population Genetics: Limitations of Single Genetic Markers
2.2. Population Genomics and Implications for Chondrichthyan Conservation and Management
2.3. Sequenced Chondrichthyan Genomes and Their Advantages
3. Genomic Assessment Techniques for Chondrichthyan Taxonomic and Population Assessments
3.1. Sub-Sampling Chondrichthyan Genomes Through Targeted Approaches
3.2. Genome Skimming: A Non-Targeted Approach to Sub-Sampling Chondrichthyan Genomes
4. Microbiome as a Health Monitoring Tool for Chondrichthyes
4.1. 16S rRNA and Shot-Gun Metagenomics to Study Chondrichthyan Microbiomes
4.2. Microbiomes in a Changing Environment
4.3. Metagenomics to Obtain Host Genomes
5. Environmental DNA (eDNA)—To Monitor the Elusive Ocean Predator
6. Taking Genomics to the Field
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Edwards, R.A.; Haggerty, J.M.; Cassman, N.; Busch, J.C.; Aguinaldo, K.; Chinta, S.; Vaughn, M.H.; Morey, R.; Harkins, T.T.; Teiling, C.; et al. Microbes, metagenomes and marine mammals: enabling the next generation of scientist to enter the genomic era. BMC Genom. 2013, 14, 600. [Google Scholar] [CrossRef] [PubMed]
- Ellegren, H. Genome sequencing and population genomics in non-model organisms. Trends Ecol. Evol. 2014, 29, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Guyer, M.S.; Collins, F.S. The Human Genome Project and the future of medicine. Am. J. Dis. Child. 1993, 147, 1145–1152. [Google Scholar] [CrossRef] [PubMed]
- Haggerty, J.M.; Dinsdale, E.A. Distinct biogeographical patterns of marine bacterial taxonomy and functional genes. Glob. Ecol. Biogeogr. 2017, 26, 177–190. [Google Scholar] [CrossRef]
- Ma, D.; Liu, F. Genome Editing and Its Applications in Model Organisms. Geno. Proteomics Bioinform. 2015, 13, 336–344. [Google Scholar] [CrossRef]
- Minich, J.J.; Morris, M.M.; Brown, M.; Doane, M.; Edwards, M.S.; Michael, T.P.; Dinsdale, E.A. Elevated temperature drives kelp microbiome dysbiosis, while elevated carbon dioxide induces water microbiome disruption. PLOS ONE 2018, 13, e0192772. [Google Scholar] [CrossRef]
- Turnbaugh, P.J.; Ley, R.E.; Hamady, M.; Fraser-Liggett, C.; Knight, R.; Gordon, J.I. The human microbiome project: exploring the microbial part of ourselves in a changing world. Nature 2007, 449, 804–810. [Google Scholar] [CrossRef]
- Menegon, M.; Cantaloni, C.; Rodriguez-Prieto, A.; Centomo, C.; Abdelfattah, A.; Rossato, M.; Bernardi, M.; Xumerle, L.; Loader, S.; Delledonne, M. On site DNA barcoding by nanopore sequencing. PLoS ONE 2017, 12, e0184741. [Google Scholar] [CrossRef]
- Morris, M.M.; Haggerty, J.M.; Papudeshi, B.N.; Vega, A.A.; Edwards, M.S.; Dinsdale, E.A. Nearshore Pelagic Microbial Community Abundance Affects Recruitment Success of Giant Kelp, Macrocystis pyrifera. Front. Microbiol. 2016, 7, 1800. [Google Scholar] [CrossRef]
- Richter, S.; Schwarz, F.; Hering, L.; Böggemann, M.; Bleidorn, C. The Utility of Genome Skimming for Phylogenomic Analyses as Demonstrated for Glycerid Relationships (Annelida, Glyceridae). Genome Biol. Evol. 2015, 7, 3443–3462. [Google Scholar] [CrossRef] [PubMed]
- Ripma, L.A.; Simpson, M.G.; Hasenstab-Lehman, K. Geneious! Simplified genome skimming methods for phylogenetic systematic studies: A case study in Oreocarya (Boraginaceae). Appl. Plant Sci. 2014, 2, 1400062. [Google Scholar] [CrossRef] [PubMed]
- Kriwet, J.; Witzmann, F.; Klug, S.; Heidtke, U.H.J. First direct evidence of a vertebrate three-level trophic chain in the fossil record. Proc. R. Soc. B Biol. Sci. 2008, 275, 181–186. [Google Scholar] [CrossRef]
- Stein, R.W.; Mull, C.G.; Kuhn, T.S.; Aschliman, N.C.; Davidson, L.N.K.; Joy, J.B.; Smith, G.J.; Dulvy, N.K.; Mooers, A.O. Global priorities for conserving the evolutionary history of sharks, rays and chimaeras. Nat. Ecol. Evol. 2018, 2, 288–298. [Google Scholar] [CrossRef]
- Marra, N.J.; Stanhope, M.J.; Jue, N.K.; Wang, M.; Sun, Q.; Bitar, P.P.; Richards, V.P.; Komissarov, A.; Rayko, M.; Kliver, S.; et al. White shark genome reveals ancient elasmobranch adaptations associated with wound healing and the maintenance of genome stability. Proc. Natl. Acad. Sci. USA 2019, 116, 4446–4455. [Google Scholar] [CrossRef] [PubMed]
- Reif, W.-E. Wound healing in Sharks: Form and arrangement of repair scales. Zoomorphologie 1978, 90, 101–111. [Google Scholar] [CrossRef]
- Chin, A.; Mourier, J.; Rummer, J.L. Blacktip reef sharks (Carcharhinus melanopterus) show high capacity for wound healing and recovery following injury. Conserv. Physiol. 2015, 3. [Google Scholar] [CrossRef] [PubMed]
- Domingues, R.R.; Hilsdorf, A.W.S.; Gadig, O.B.F. The importance of considering genetic diversity in shark and ray conservation policies. Conserv. Genet. 2018, 19, 501–525. [Google Scholar] [CrossRef]
- Venkatesh, B.; Lee, A.P.; Ravi, V.; Maurya, A.K.; Lian, M.M.; Swann, J.B.; Ohta, Y.; Flajnik, M.F.; Sutoh, Y.; Kasahara, M.; et al. Elephant shark genome provides unique insights into gnathostome evolution. Nature 2014, 505, 174–179. [Google Scholar] [CrossRef]
- Dulvy, N.K.; Fowler, S.L.; Musick, J.A.; Cavanagh, R.D.; Kyne, P.M.; Harrison, L.R.; Carlson, J.K.; Davidson, L.N.; Fordham, S.V.; Francis, M.P.; et al. Extinction risk and conservation of the world’s sharks and rays. eLife 2014, 3, e00590. [Google Scholar] [CrossRef]
- García, V.B.; Lucifora, L.O.; Myers, R.A. The importance of habitat and life history to extinction risk in sharks, skates, rays and chimaeras. Proc. R. Soc. Lond. B Biol. Sci. 2008, 275, 83–89. [Google Scholar] [CrossRef]
- Taguchi, M.; King, J.R.; Wetklo, M.; Withler, R.E.; Yokawa, K. Population Genetic Structure and Demographic History of Pacific Blue Sharks (Prionace glauca) Inferred from Mitochondrial DNA Analysis—Dimensions. Available online: https://app.dimensions.ai/details/publication/pub.1035226050 (accessed on 24 September 2018).
- Manuzzi, A.; Zane, L.; Muñoz-Merida, A.; Griffiths, A.M.; Veríssimo, A. Population genomics and phylogeography of a benthic coastal shark (Scyliorhinus canicula) using 2b-RAD single nucleotide polymorphisms. Biol. J. Linn. Soc. 2019, 126, 289–303. [Google Scholar] [CrossRef]
- Gallagher, A.J.; Hammerschlag, N.; Shiffman, D.S.; Giery, S.T. Evolved for Extinction: The Cost and Conservation Implications of Specialization in Hammerhead Sharks. BioScience 2014, 64, 619–624. [Google Scholar] [CrossRef]
- Stevens, J. The effects of fishing on sharks, rays, and chimaeras (chondrichthyans), and the implications for marine ecosystems. ICES J. Mar. Sci. 2000, 57, 476–494. [Google Scholar] [CrossRef]
- Bernatchez, L.; Wellenreuther, M.; Araneda, C.; Ashton, D.T.; Barth, J.M.I.; Beacham, T.D.; Maes, G.E.; Martinsohn, J.T.; Miller, K.M.; Naish, K.A.; et al. Harnessing the Power of Genomics to Secure the Future of Seafood. Trends Ecol. Evol. 2017, 32, 665–680. [Google Scholar] [CrossRef]
- Carvalho, G.R.; Hauser, L.; Martinsohn, J.; Naish, K. Fish, genes and genomes: contributions to ecology, evolution and management. J. Fish Biol. 2016, 89, 2471–2478. [Google Scholar] [CrossRef]
- Ungerer, M.C.; Johnson, L.C.; Herman, M.A. Ecological genomics: Understanding gene and genome function in the natural environment. Heredity 2008, 100, 178–183. [Google Scholar] [CrossRef]
- Dudgeon, C.L.; Blower, D.C.; Broderick, D.; Giles, J.L.; Holmes, B.J.; Kashiwagi, T.; Krück, N.C.; Morgan, J.A.T.; Tillett , B.J.; Ovenden, J.R. A review of the application of molecular genetics for fisheries management and conservation of sharks and rays. J. Fish Biol. 2012, 80, 1789–1843. [Google Scholar] [CrossRef]
- Hebert, P.D.N.; Cywinska, A.; Ball, S.L.; de Waard, J.R. Biological identifications through DNA barcodes. Proc. R. Soc. B Biol. Sci. 2003, 270, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Bakker, J.; Wangensteen, O.S.; Chapman, D.D.; Boussarie, G.; Buddo, D.; Guttridge, T.L.; Hertler, H.; Mouillot, D.; Vigliola, L.; Mariani, S. Environmental DNA reveals tropical shark diversity in contrasting levels of anthropogenic impact. Sci. Rep. 2017, 7, 16886. [Google Scholar] [CrossRef] [PubMed]
- Bineesh, K.K.; Gopalakrishnan, A.; Akhilesh, K.V.; Sajeela, K.A.; Abdussamad, E.M.; Pillai, N.G.K.; Basheer, V.S.; Jena, J.K.; Ward, R.D. DNA barcoding reveals species composition of sharks and rays in the Indian commercial fishery. Mitochondrial DNA Part DNA Mapp. Seq. Anal. 2016, 28, 458–472. [Google Scholar] [CrossRef]
- Chuang, P.-S.; Hung, T.-C.; Chang, H.-A.; Huang, C.-K.; Shiao, J.-C. The Species and Origin of Shark Fins in Taiwan’s Fishing Ports, Markets, and Customs Detention: A DNA Barcoding Analysis. PLoS ONE 2016, 11, e0147290. [Google Scholar] [CrossRef] [PubMed]
- Steinke, D.; Bernard, A.M.; Horn, R.L.; Hilton, P.; Hanner, R.; Shivji, M.S. DNA analysis of traded shark fins and mobulid gill plates reveals a high proportion of species of conservation concern. Sci. Rep. 2017, 7, 9505. [Google Scholar] [CrossRef]
- Henderson, A.C.; Reeve, A.J.; Jabado, R.W.; Naylor, G.J.P. Taxonomic assessment of sharks, rays and guitarfishes (Chondrichthyes: Elasmobranchii) from south-eastern Arabia, using the NADH dehydrogenase subunit 2 (NADH2) gene. Zool. J. Linn. Soc. 2016, 176, 399–442. [Google Scholar] [CrossRef]
- Naylor, G.J.P.; Caira, J.N.; Jensen, K.; Rosana, K.A.M.; White, W.T.; Last, P.R. A DNA sequence-based approach to the identification of shark and ray species and its implications for global elasmobranch diversity and parasitology. (Bulletin of the American Museum of Natural History, no. 367). DNA Identif. Sharks Rays 2012. Available online: http://digitallibrary.amnh.org/handle/2246/6183 (accessed on 18 March 2019).
- Boomer, J.J.; Peddemors, V.; Stow, A.J. Genetic data show that Carcharhinus tilstoni is not confined to the tropics, highlighting the importance of a multifaceted approach to species identification. J. Fish Biol. 2010, 77, 1165–1172. [Google Scholar] [CrossRef]
- Morgan, J.A.T.; Harry, A.V.; Welch, D.J.; Street, R.; White, J.; Geraghty, P.T.; Macbeth, W.G.; Tobin, A.; Simpfendorfer, C.A.; Ovenden, J.R. Detection of interspecies hybridisation in Chondrichthyes: hybrids and hybrid offspring between Australian (Carcharhinus tilstoni) and common (C. limbatus) blacktip shark found in an Australian fishery. Conserv. Genet. 2012, 13, 455–463. [Google Scholar] [CrossRef]
- Rodrigues-Filho, L.F.d.S.; da Rocha, T.C.; do Rêgo, P.S.; Schneider, H.; Sampaio, I.; Vallinoto, M. Identification and phylogenetic inferences on stocks of sharks affected by the fishing industry off the Northern coast of Brazil. Genet. Mol. Biol. 2009, 32, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Pinhal, D.; Shivji, M.S.; Nachtigall, P.G.; Chapman, D.D.; Martins, C. A streamlined DNA tool for global identification of heavily exploited coastal shark species (genus Rhizoprionodon). PLoS ONE 2012, 7, e34797. [Google Scholar] [CrossRef] [PubMed]
- Moore, A.B.M.; White, W.T.; Ward, R.D.; Naylor, G.J.P.; Peirce, R. Rediscovery and redescription of the smoothtooth blacktip shark, Carcharhinus leiodon (Carcharhinidae), from Kuwait, with notes on its possible conservation status. Mar. Freshw. Res. 2011, 62, 528. [Google Scholar] [CrossRef]
- Ward, R.D.; Zemlak, T.S.; Innes, B.H.; Last, P.R.; Hebert, P.D. DNA barcoding Australia’s fish species. Philos. Trans. R. Soc. B Biol. Sci. 2005, 360, 1847–1857. [Google Scholar] [CrossRef]
- Johri, S.; Solanki, J.; Cantu, V.A.; Fellows, S.R.; Edwards, R.A.; Moreno, I.; Vyas, A.; Dinsdale, E.A. ‘Genome skimming’ with the MinION hand-held sequencer identifies CITES-listed shark species in India’s exports market. Sci. Rep. 2019, 9, 4476. [Google Scholar] [CrossRef] [PubMed]
- Naylor, G.J.; Ryburn, J.A.; Fedrigo, O.; Lopez, J.A. Phylogenetic relationships among the major lineages of modern elasmobranchs. Reprod. Biol. Phylogeny 2005, 3, 25. [Google Scholar]
- Haasl, R.J.; Payseur, B.A. Microsatellites as Targets of Natural Selection. Mol. Biol. Evol. 2013, 30, 285–298. [Google Scholar] [CrossRef]
- Hoelzel, A.R.; Shivji, M.S.; Magnussen, J.; Francis, M.P. Low worldwide genetic diversity in the basking shark (Cetorhinus maximus). Biol. Lett. 2006, 2, 639–642. [Google Scholar] [CrossRef] [PubMed]
- Castro, A.L.F. Use of molecular tools on surveys of genetic variation and population structure in three species of sharks. Ph.D. Thesis, University of South Florida, Tampa, FL, USA, 2009. Available online: https://scholarcommons.usf.edu/etd/1893 (accessed on 18 October 2018).
- Karl, S.A.; Castro, A.L.F.; Garla, R.C. Population genetics of the nurse shark (Ginglymostoma cirratum) in the western. Atlantic. Mar. Biol. 2012, 159, 489–498. [Google Scholar] [CrossRef]
- Pardini, A.T.; Jones, C.; Noble, L.; Kreiser, B.; Malcolm, H.; Bruce, B.D.; Stevens, J.D.; Cliff, G.; Scholl, M.; Francis, M.; et al. Sex-biased dispersal of great white sharks - In some respects, these sharks behave more like whales and dolphins than other fish. Nature 2001, 412, 139–140. [Google Scholar] [CrossRef]
- Gubili, C.; Bilgin, R.; Kalkan, E.; Karhan, U.; Jones, C.S.; Sims, D.W.; Kabasakal, H.; Martin, A.; Noble, L.R. Antipodean white sharks on a Mediterranean walkabout? Historical dispersal leads to genetic discontinuity and an endangered anomalous population. Proc. R. Soc. Lond. B Biol. Sci. 2011, 278, 1679–1686. [Google Scholar] [CrossRef]
- Keeney, D.B.; Heist, E.J. Worldwide phylogeography of the blacktip shark (Carcharhinus limbatus) inferred from mitochondrial DNA reveals isolation of western Atlantic populations coupled with recent Pacific dispersal. Mol. Ecol. 2006, 15, 3669–3679. [Google Scholar] [CrossRef] [PubMed]
- Karl, S.; Castro, A.L.F.; López, J.; Charvet, P.; Burgess, G. Phylogeography and conservation of the bull shark (Carcharhinus leucas) inferred from mitochondrial and microsatellite DNA. Conserv. Genet. 2011, 12, 371–382. [Google Scholar] [CrossRef]
- Tillett, B.J.; Meekan, M.G.; Broderick, D.; Field, I.C.; Cliff, G.; Ovenden, J.R. Pleistocene isolation, secondary introgression and restricted contemporary gene flow in the pig-eye shark, Carcharhinus amboinensis across northern Australia. Conserv. Genet. 2012, 13, 99–115. [Google Scholar] [CrossRef]
- Ovenden, J.R.; Morgan, J.A.T.; Street, R.; Tobin, A.; Simpfendorfer, C.; Macbeth, W.; Welch, D. Negligible evidence for regional genetic population structure for two shark species Rhizoprionodon acutus (Rüppell, 1837) and Sphyrna lewini (Griffith & Smith, 1834) with contrasting biology. Mar. Biol. 2011, 158, 1497–1509. [Google Scholar]
- Veríssimo, A.; McDowell, J.R.; Graves, J.E. Population structure of a deep-water squaloid shark, the Portuguese dogfish (Centroscymnus coelolepis). ICES J. Mar. Sci. 2011, 68, 555–563. [Google Scholar] [CrossRef]
- Veríssimo, A.; McDowell, J.R.; Graves, J.E. Global population structure of the spiny dogfish Squalus acanthias, a temperate shark with an antitropical distribution. Mol. Ecol. 2010, 19, 1651–1662. [Google Scholar] [CrossRef]
- Bester-van der Merwe, A.E.; Bitalo, D.; Cuevas, J.M.; Ovenden, J.; Hernández, S.; da Silva, C.; McCord, M.; Roodt-Wilding, R. Population genetics of Southern Hemisphere tope shark (Galeorhinus galeus): Intercontinental divergence and constrained gene flow at different geographical scales. PLOS ONE 2017, 12, e0184481. [Google Scholar] [CrossRef]
- Crandall, K.; Bininda-Emonds, O.; Mace, G.; Wayne, R.K. Considering evolutionary process in conservation biology. Trends Ecol. Evol. 2000, 15, 290–295. [Google Scholar] [CrossRef]
- Frankel, O.H. Genetic conservation: our evolutionary responsibility. Genetics 1974, 78, 53–65. [Google Scholar] [PubMed]
- Ryder, O. Species conservation and systematics: the dilemma of subspecies. Trends Ecol. Evol. 1986, 1, 9–10. [Google Scholar]
- Brumfield, R.T.; Beerli, P.; Nickerson, D.A.; Edwards, S.V. The utility of single nucleotide polymorphisms in inferences of population history. Trends Ecol. Evol. 2003, 18, 249–256. [Google Scholar] [CrossRef]
- Pirog, A.; Blaison, A.; Jaquemet, S.; Soria, M.; Magalon, H. Isolation and characterization of 20 microsatellite markers from Carcharhinus leucas (bull shark) and cross-amplification in Galeocerdo cuvier (tiger shark), Carcharhinus obscurus (dusky shark) and Carcharhinus plumbeus (sandbar shark). Conserv. Genet. Resour. 2015, 7, 121–124. [Google Scholar] [CrossRef]
- Mendes, N.J.; Cruz, V.P.; Mendonça, F.F.; Pardo, B.G.; Coelho, R.; Ashikaga, F.Y.; Camargo, S.M.; Martínez, P.; Oliveira, C.; Santos, M.N.; et al. Microsatellite loci in the oceanic whitetip shark and cross-species amplification using pyrosequencing technology. Conserv. Genet. Resour. 2015, 7, 585–589. [Google Scholar] [CrossRef]
- Mendes, N.J.; Cruz, V.P.; Ashikaga, F.Y.; Camargo, S.M.; Oliveira, C.; Piercy, A.N.; Burgess, G.H.; Coelho, R.; Santos, M.N.; Mendonça, F.F.; et al. Microsatellite loci in the tiger shark and cross-species amplification using pyrosequencing technology. PeerJ 2016, 4, e2205. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Hofreiter, M.; Straube, N.; Corrigan, S.; Naylor, G.J.P. Capturing protein-coding genes across highly divergent species. BioTechniques 2013, 54, 321–326. [Google Scholar] [CrossRef]
- Momigliano, P.; Harcourt, R.; Robbins, W.D.; Jaiteh, V.; Mahardika, G.N.; Sembiring, A.; Stow, A. Genetic structure and signatures of selection in grey reef sharks (Carcharhinus amblyrhynchos). Heredity 2017, 119, 142–153. [Google Scholar] [CrossRef]
- Pazmiño, D.A.; Maes, G.E.; Simpfendorfer, C.A.; Salinas-de-León, P.; Herwerden, L. Van Genome-wide SNPs reveal low effective population size within confined management units of the highly vagile Galapagos shark (Carcharhinus galapagensis). Conserv. Genet. 2017, 18, 1151–1163. [Google Scholar] [CrossRef]
- Planes, S.; Fauvelot, C. Isolation by distance and vicariance drive genetic structure of a coral reef fish in the Pacific Ocean. Evol. Int. J. Org. Evol. 2002, 56, 378–399. [Google Scholar] [CrossRef]
- Pazmiño, D.; Maes, G.; Green, M.; Simpfendorfer, C.; Hoyos, M.; Duffy, C.; Meyer, C.; Kerwath, S.; Salinas-de-Leon, P.; Herwerden, L. Strong trans-Pacific break and local conservation units in the Galapagos shark (Carcharhinus galapagensis) revealed by genome-wide cytonuclear markers. Heredity 2018, 120. [Google Scholar] [CrossRef] [PubMed]
- Portnoy, D.; Puritz, J.; Hollenbeck, C.M.; Gelsleichter, J.; Chapman, D.; Gold, J. Selection and sex-biased dispersal in a coastal shark: The influence of philopatry on adaptive variation. Mol. Ecol. 2015, 24, 5885. [Google Scholar] [CrossRef] [PubMed]
- Read, T.D.; Petit, R.A.; Joseph, S.J.; Alam, M.T.; Weil, M.R.; Ahmad, M.; Bhimani, R.; Vuong, J.S.; Haase, C.P.; Webb, D.H.; et al. Draft sequencing and assembly of the genome of the world’s largest fish, the whale shark: Rhincodon typus Smith 1828. BMC Genom. 2017, 18, 532. [Google Scholar]
- Hara, Y.; Yamaguchi, K.; Onimaru, K.; Kadota, M.; Koyanagi, M.; Keeley, S.D.; Tatsumi, K.; Tanaka, K.; Motone, F.; Kageyama, Y.; et al. Shark genomes provide insights into elasmobranch evolution and the origin of vertebrates. Nat. Ecol. Evol. 2018, 2, 1761. [Google Scholar] [CrossRef]
- Dooley, H.; Flajnik, M.F. Shark immunity bites back: Affinity maturation and memory response in the nurse shark, Ginglymostoma cirratum. Eur. J. Immunol. 2015, 35, 936–945. [Google Scholar] [CrossRef]
- Bernal, M.A.; Sinai, N.L.; Rocha, C.; Gaither, M.R.; Dunker, F.; Rocha, L.A. Long-term sperm storage in the brownbanded bamboo shark Chiloscyllium punctatum. J. Fish Biol. 2014, 86, 1171–1176. [Google Scholar] [CrossRef] [PubMed]
- Delser, P.M.; Corrigan, S.; Hale, M.; Li, C.; Veuille, M.; Planes, S.; Naylor, G.; Mona, S. Population genomics of C. melanopterus using target gene capture data: demographic inferences and conservation perspectives. Sci. Rep. 2016, 6, 33753. [Google Scholar] [CrossRef]
- Etter, P.D.; Bassham, S.; Hohenlohe, P.A.; Johnson, E.A.; Cresko, W.A. SNP discovery and genotyping for evolutionary genetics using RAD sequencing. Methods Mol. Biol. 2011, 772, 157–178. [Google Scholar] [PubMed]
- Maduna, S.N.; Rossouw, C.; Slabbert, R.; Wintner, S.P.; da Silva, C.; Bester-van der Merwe, A.E. New polymorphic microsatellite loci revealed for the dusky shark Carcharhinus obscurus through Ion Proton double-digest RAD sequencing. Mol. Biol. Rep. 2018. [Google Scholar] [CrossRef]
- Galván-Tirado, C.; Hinojosa-Alvarez, S.; Diaz-Jaimes, P.; Marcet-Houben, M.; García-De-León, F.J. The complete mitochondrial DNA of the silky shark (Carcharhinus falciformis). Mitochondrial DNA Part A Mapp. Seq. Anal. 2016, 27, 157–158. [Google Scholar] [CrossRef]
- Berger, B.A.; Han, J.; Sessa, E.B.; Gardner, A.G.; Shepherd, K.A.; Ricigliano, V.A.; Jabaily, R.S.; Howarth, D.G. The Unexpected Depths of Genome-Skimming Data: A Case Study Examining Goodeniaceae Floral Symmetry Genes. Appl. Plant. Sci. 2017, 5, 1700042. [Google Scholar] [CrossRef]
- Beattie, G.A. Metabolic coupling on roots. Nat. Microbiol. 2018, 3, 396–397. [Google Scholar] [CrossRef]
- Cassman, N.; Prieto-Davó, A.; Walsh, K.; Silva, G.G.Z.; Angly, F.; Akhter, S.; Barott, K.; Busch, J.; McDole, T.; Haggerty, J.M.; et al. Oxygen minimum zones harbour novel viral communities with low diversity. Environ. Microbiol. 2012, 14, 3043–3065. [Google Scholar] [CrossRef] [PubMed]
- Jeffries, T.C.; Seymour, J.R.; Gilbert, J.A.; Dinsdale, E.A.; Newton, K.; Leterme, S.S.C.; Roudnew, B.; Smith, R.J.; Seuront, L.; Mitchell, J.G. Substrate Type Determines Metagenomic Profiles from Diverse Chemical Habitats. PLOS ONE 2011, 6, e25173. [Google Scholar] [CrossRef]
- Dinsdale, E.A.; Edwards, R.A.; Hall, D.; Angly, F.; Breitbart, M.; Brulc, J.M.; Furlan, M.; Desnues, C.; Haynes, M.; Li, L.; et al. Functional metagenomic profiling of nine biomes. Nature 2008, 452, 629–632. [Google Scholar] [CrossRef]
- Walsh, K.; Haggerty, J.M.; Doane, M.P.; Hansen, J.J.; Morris, M.M.; Moreira, A.P.B.; de Oliveira, L.; Leomil, L.; Garcia, G.D.; Thompson, F.; et al. Aura-biomes are present in the water layer above coral reef benthic macro-organisms. PeerJ 2017, 5, e3666. [Google Scholar] [CrossRef] [PubMed]
- Burke, C.; Steinberg, P.; Rusch, D.; Kjelleberg, S.; Thomas, T. Bacterial community assembly based on functional genes rather than species. Proc. Natl. Acad. Sci. USA 2011, 108, 14288–14293. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.; Reynolds, D.; Liu, M.; Stark, M.; Kjelleberg, S.; Webster, N.S.; Thomas, T. Functional equivalence and evolutionary convergence in complex communities of microbial sponge symbionts. Proc. Natl. Acad. Sci. USA 2012, 109, E1878–E1887. [Google Scholar] [CrossRef] [PubMed]
- Larsen, A.; Tao, Z.; Bullard, S.A.; Arias, C.R. Diversity of the skin microbiota of fishes: evidence for host species specificity. FEMS Microbiol. Ecol. 2013, 85, 483–494. [Google Scholar] [CrossRef] [PubMed]
- McKenzie, V.J.; Bowers, R.M.; Fierer, N.; Knight, R.; Lauber, C.L. Co-habiting amphibian species harbor unique skin bacterial communities in wild populations. ISME J. 2012, 6, 588–596. [Google Scholar] [CrossRef] [PubMed]
- Rohwer, F.; Seguritan, V.; Azam, F.; Knowlton, N. Diversity and distribution of coral-associated bacteria. Mar. Ecol. Progr. Ser. 2002, 243, 1–10. [Google Scholar] [CrossRef]
- Sison-Mangus, M.P.; Jiang, S.; Kudela, R.M.; Mehic, S. Phytoplankton-Associated Bacterial Community Composition and Succession during Toxic Diatom Bloom and Non-Bloom Events. Front. Microbiol. 2016, 7. [Google Scholar] [CrossRef]
- Sison-Mangus, M.P.; Jiang, S.; Tran, K.N.; Kudela, R.M. Host-specific adaptation governs the interaction of the marine diatom, Pseudo-nitzschia and their microbiota. ISME J. 2014, 8, 63–76. [Google Scholar] [CrossRef]
- Consortium, T.H.M.P.; Huttenhower, C.; Gevers, D.; Knight, R.; Abubucker, S.; Badger, J.H.; Chinwalla, A.T.; Creasy, H.H.; Earl, A.M.; FitzGerald, M.G.; et al. Structure, function and diversity of the healthy human microbiome. Nature 2012, 486, 207–214. [Google Scholar]
- Hooper, L.V.; Littman, D.R.; Macpherson, A.J. Interactions between the microbiota and the immune system. Science 2012, 336, 1268–1273. [Google Scholar] [CrossRef]
- Kau, A.L.; Ahern, P.P.; Griffin, N.W.; Goodman, A.L.; Gordon, J.I. Human nutrition, the gut microbiome and the immune system. Nature 2011, 474, 327–336. [Google Scholar] [CrossRef]
- Joint, I.; Tait, K.; Callow, M.E.; Callow, J.A.; Milton, D.; Williams, P.; Cámara, M. Cell-to-Cell Communication Across the Prokaryote-Eukaryote Boundary. Science 2002, 298, 1207. [Google Scholar] [CrossRef]
- Fox, C.; Eichelberger, K. Maternal microbiome and pregnancy outcomes. Fertil. Steril. 2015, 104, 1358–1363. [Google Scholar] [CrossRef]
- Busch, J.; Nascimento, J.R.; Magalhães, A.C.R.; Dutilh, B.E.; Dinsdale, E. Copper tolerance and distribution of epibiotic bacteria associated with giant kelp Macrocystis pyrifera in southern California. Ecotoxicology 2015, 24, 1131–1140. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Nichols, R.G.; Correll, J.; Murray, I.A.; Tanaka, N.; Smith, P.B.; Hubbard, T.D.; Sebastian, A.; Albert, I.; Hatzakis, E.; et al. Persistent Organic Pollutants Modify Gut Microbiota–Host Metabolic Homeostasis in Mice Through Aryl Hydrocarbon Receptor Activation. Environ. Health Perspect. 2015, 123, 679–688. [Google Scholar] [CrossRef]
- Amato, K.R.; Yeoman, C.J.; Kent, A.; Righini, N.; Carbonero, F.; Estrada, A.; Gaskins, H.R.; Stumpf, R.M.; Yildirim, S.; Torralba, M.; et al. Habitat degradation impacts black howler monkey (Alouatta pigra) gastrointestinal microbiomes. ISME J. 2013, 7, 1344–1353. [Google Scholar] [CrossRef] [PubMed]
- Doane, M.P.; Haggerty, J.M.; Kacev, D.; Papudeshi, B.; Dinsdale, E.A. The skin microbiome of the common thresher shark (Alopias vulpinus) has low taxonomic and gene function β-diversity. Environ. Microbiol. Rep. 2017, 9, 357–373. [Google Scholar] [CrossRef]
- Apprill, A.; Miller, C.A.; Moore, M.J.; Durban, J.W.; Fearnbach, H.; Barrett-Lennard, L.G. Extensive Core Microbiome in Drone-Captured Whale Blow Supports a Framework for Health Monitoring. mSystems 2017, 2, e00119-17. [Google Scholar] [CrossRef]
- Nussbaumer, A.D.; Fisher, C.R.; Bright, M. Horizontal endosymbiont transmission in hydrothermal vent tubeworms. Nature 2006, 441, 345–348. [Google Scholar] [CrossRef] [PubMed]
- Sharp, K.H.; Eam, B.; Faulkner, D.J.; Haygood, M.G. Vertical Transmission of Diverse Microbes in the Tropical Sponge Corticium sp. Appl. Environ. Microbiol. 2007, 73, 622–629. [Google Scholar]
- Zeng, Q.; Wu, S.; Sukumaran, J.; Rodrigo, A. Models of microbiome evolution incorporating host and microbial selection. Microbiome 2017, 5, 127. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Q.; Rodrigo, A. Neutral models of short-term microbiome dynamics with host subpopulation structure and migration limitation. Microbiome 2018, 6, 80. [Google Scholar] [CrossRef] [PubMed]
- Egan, S.; Gardiner, M. Microbial Dysbiosis: Rethinking Disease in Marine Ecosystems. Front. Microbiol. 2016, 7, 991. [Google Scholar] [CrossRef]
- Cavalcanti, G.S.; Shukla, P.; Morris, M.; Ribeiro, B.; Foley, M.; Doane, M.P.; Thompson, C.C.; Edwards, M.S.; Dinsdale, E.A.; Thompson, F.L. Rhodoliths holobionts in a changing ocean: host-microbes interactions mediate coralline algae resilience under ocean acidification. BMC Genom. 2018, 19, 701. [Google Scholar] [CrossRef]
- Janssens, Y.; Nielandt, J.; Bronselaer, A.; Debunne, N.; Verbeke, F.; Wynendaele, E.; Van Immerseel, F.; Vandewynckel, Y.-P.; De Tré, G.; De Spiegeleer, B. Disbiome database: linking the microbiome to disease. BMC Microbiol. 2018, 18, 50. [Google Scholar] [CrossRef] [PubMed]
- Liang, D.; Leung, R.K.-K.; Guan, W.; Au, W.W. Involvement of gut microbiome in human health and disease: Brief overview, knowledge gaps and research opportunities. Gut Pathog. 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Apprill, A.; Robbins, J.; Eren, A.M.; Pack, A.A.; Reveillaud, J.; Mattila, D.; Moore, M.; Niemeyer, M.; Moore, K.M.T.; Mincer, T.J. Humpback Whale Populations Share a Core Skin Bacterial Community: Towards a Health Index for Marine Mammals? PLoS ONE 2014, 9, e90785. [Google Scholar] [CrossRef] [PubMed]
- Colston, T.; Jackson, C. Invited Review: Microbiome Evolution Along Divergent Branches of the Vertebrate Tree of Life: What’s Known and Unknown. Mol. Ecol. 2016, 25. [Google Scholar] [CrossRef]
- Givens, C.E.; Ransom, B.; Bano, N.; Hollibaugh, J.T. Comparison of the gut microbiomes of 12 bony fish and 3 shark species. Mar. Ecol. Prog. Ser. 2015, 518, 209–223. [Google Scholar] [CrossRef]
- Kearns, P.J.; Bowen, J.L.; Tlusty, M.F. The skin microbiome of cow-nose rays (Rhinoptera bonasus) in an aquarium touch-tank exhibit. Zoo Biol. 2017, 36, 226–230. [Google Scholar] [CrossRef] [PubMed]
- Sherrill-Mix, S.; McCormick, K.; Lauder, A.; Bailey, A.; Zimmerman, L.; Li, Y.; Django, J.-B.N.; Bertolani, P.; Colin, C.; Hart, J.A.; et al. Allometry and Ecology of the Bilaterian Gut Microbiome. mBio 2018, 9, e00319-18. [Google Scholar] [CrossRef]
- Barbato, M.; Kovacs, T.; Coleman, M.A.; Broadhurst, M.K.; Bruyn, M. De Metabarcoding for stomach-content analyses of Pygmy devil ray (Mobula kuhlii cf. eregoodootenkee): Comparing tissue and ethanol preservative-derived. DNA. Ecol. Evol. 2019, 9, 2678–2687. [Google Scholar] [PubMed]
- Johny, T.K.; Saidumohamed, B.E.; Sasidharan, R.S.; Bhat, S.G. Metabarcoding data of bacterial diversity of the deep sea shark, Centroscyllium fabricii. Data Brief 2018, 21, 1029–1032. [Google Scholar] [CrossRef] [PubMed]
- Retallack, H.; Okihiro, M.S.; Britton, E.; Van Sommeran, S.; DeRisi, J.L. Metagenomic Next-Generation Sequencing Reveals Miamiensis avidus (Ciliophora: Scuticociliatida) in the 2017 Epizootic of Leopard Sharks (Triakis semifasciata) in San Francisco Bay, California, USA. J. Wildl. Dis. 2018, 55. [Google Scholar] [CrossRef] [PubMed]
- Doane, M.P.; Kacev, D.; Harrington, S.; Levi, K.; Pande, D.; Vega, A.; Dinsdale, E.A. Mitochondrial recovery from shotgun metagenome sequencing enabling phylogenetic analysis of the common thresher shark (Alopias vulpinus). Meta Gene 2018, 15, 10–15. [Google Scholar] [CrossRef]
- Kelly, L.W.; Williams, G.J.; Barott, K.L.; Carlson, C.A.; Dinsdale, E.A.; Edwards, R.A.; Haas, A.F.; Haynes, M.; Lim, Y.W.; McDole, T.; et al. Local genomic adaptation of coral reef-associated microbiomes to gradients of natural variability and anthropogenic stressors. Proc. Natl. Acad. Sci. USA 2014, 111, 10227–10232. [Google Scholar] [CrossRef] [PubMed]
- Bowman, J.P. Bioactive compound synthetic capacity and ecological significance of marine bacterial genus pseudoalteromonas. Mar. Drugs 2007, 5, 220–241. [Google Scholar] [CrossRef] [PubMed]
- Muegge, B.D.; Kuczynski, J.; Knights, D.; Clemente, J.C.; González, A.; Fontana, L.; Henrissat, B.; Knight, R.; Gordon, J.I. Diet drives convergence in gut microbiome functions across mammalian phylogeny and within humans. Science 2011, 332, 970–974. [Google Scholar] [CrossRef] [PubMed]
- Dutilh, B.E.; Schmieder, R.; Nulton, J.; Felts, B.; Salamon, P.; Edwards, R.A.; Mokili, J.L. Reference-independent comparative metagenomics using cross-assembly: crAss. Bioinformatics 2012, 28, 3225–3231. [Google Scholar] [CrossRef]
- Papudeshi, B.; Haggerty, J.M.; Doane, M.; Morris, M.M.; Walsh, K.; Beattie, D.T.; Pande, D.; Zaeri, P.; Silva, G.G.Z.; Thompson, F.; et al. Optimizing and evaluating the reconstruction of Metagenome-assembled microbial genomes. BMC Genom. 2017, 18, 915. [Google Scholar] [CrossRef]
- Parks, D.H.; Rinke, C.; Chuvochina, M.; Chaumeil, P.-A.; Woodcroft, B.J.; Evans, P.N.; Hugenholtz, P.; Tyson, G.W. Recovery of nearly 8,000 metagenome-assembled genomes substantially expands the tree of life. Nature Microbiol. 2017, 2, 1533–1542. [Google Scholar] [CrossRef]
- Alneberg, J.; Bjarnason, B.S.; de Bruijn, I.; Schirmer, M.; Quick, J.; Ijaz, U.Z.; Loman, N.J.; Andersson, A.F.; Quince, C. CONCOCT: Clustering cONtigs on COverage and ComposiTion. arXiv, 2013; arXiv:1312.4038. Available online: http://arxiv.org/abs/1312.4038) (accessed on 27 July 2018).
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- NCBI Resource Coordinators Database Resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2017, 45, D12–D17. [CrossRef] [PubMed]
- Evans, N.T.; Li, Y.; Renshaw, M.A.; Olds, B.P.; Deiner, K.; Turner, C.R.; Jerde, C.L.; Lodge, D.M.; Lamberti, G.A.; Pfrender, M.E. Fish community assessment with eDNA metabarcoding: effects of sampling design and bioinformatic filtering. Can. J. Fish. Aquat. Sci. 2017, 74, 1362–1374. [Google Scholar] [CrossRef]
- Stat, M.; John, J.; DiBattista, J.D.; Newman, S.J.; Bunce, M.; Harvey, E.S. Combined use of eDNA metabarcoding and video surveillance for the assessment of fish biodiversity: Fish Surveying. Conserv. Biol. 2019, 33, 196–205. [Google Scholar] [CrossRef] [PubMed]
- Djurhuus, A.; Port, J.; Closek, C.J.; Yamahara, K.M.; Romero-Maraccini, O.; Walz, K.R.; Goldsmith, D.B.; Michisaki, R.; Breitbart, M.; Boehm, A.B.; et al. Evaluation of Filtration and DNA Extraction Methods for Environmental DNA Biodiversity Assessments across Multiple Trophic Levels. Front. Mar. Sci. 2017, 4. [Google Scholar] [CrossRef]
- Andruszkiewicz, E.A.; Starks, H.A.; Chavez, F.P.; Sassoubre, L.M.; Block, B.A.; Boehm, A.B. Biomonitoring of marine vertebrates in Monterey Bay using eDNA metabarcoding. PLoS ONE 2017, 12, e0176343. [Google Scholar] [CrossRef] [PubMed]
- Sigsgaard, E.E.; Nielsen, I.B.; Bach, S.S.; Lorenzen, E.D.; Robinson, D.P.; Knudsen, S.W.; Pedersen, M.W.; Jaidah, M.A.; Orlando, L.; Willerslev, E.; et al. Population characteristics of a large whale shark aggregation inferred from seawater environmental DNA. Nat. Ecol. Evol. 2016, 1, s41559-016. [Google Scholar] [CrossRef] [PubMed]
- Lafferty, K.D.; Benesh, K.C.; Mahon, A.R.; Jerde, C.L.; Lowe, C.G. Detecting Southern California’s White Sharks With Environmental DNA. Front. Mar. Sci. 2018, 5. [Google Scholar] [CrossRef]
- Fields, A.T.; Abercrombie, D.L.; Eng, R.; Feldheim, K.; Chapman, D.D. A Novel Mini-DNA Barcoding Assay to Identify Processed Fins from Internationally Protected Shark Species. PLoS ONE 2015, 10, e0114844. [Google Scholar] [CrossRef]
- Boussarie, G.; Bakker, J.; Wangensteen, O.S.; Mariani, S.; Bonnin, L.; Juhel, J.-B.; Kiszka, J.J.; Kulbicki, M.; Manel, S.; Robbins, W.D.; et al. Environmental DNA illuminates the dark diversity of sharks. Sci. Adv. 2018, 4, eaap9661. [Google Scholar] [CrossRef]
- Dewar, H.; Eguchi, T.; Hyde, J.; Kinzey, D.; Kohin, S.; Moore, J.; Taylor, B.; Vetter, R. Status Review of the Northeastern Pacific Population of White Sharks (Carcharodon carcharias) under the Endangered Species Act. Available online: https://swfsc.noaa.gov/publications/TM/SWFSC/NOAA-TM-NMFS-SWFSC-523.pdf (accessed on 23 March 2019).
- Cape Cod’s Gray Seal and White Shark Problem Is Anything but Black-and-White. Available online: https://www.nrdc.org/onearth/cape-cods-gray-seal-and-white-shark-problem-anything-black-and-white (accessed on 18 March 2019).
- Laver, T.; Harrison, J.; O’Neill, P.A.; Moore, K.; Farbos, A.; Paszkiewicz, K.; Studholme, D.J. Assessing the performance of the Oxford Nanopore Technologies MinION. Biomol. Detect. Quantif. 2015, 3, 1–8. [Google Scholar] [CrossRef]
- Jansen, H.J.; Liem, M.; Jong-Raadsen, S.A.; Dufour, S.; Weltzien, F.-A.; Swinkels, W.; Koelewijn, A.; Palstra, A.P.; Pelster, B.; Spaink, H.P.; et al. Rapid de novo assembly of the European eel genome from nanopore sequencing reads. Sci. Rep. 2017, 7, 7213. [Google Scholar] [CrossRef] [PubMed]
- Minei, R.; Hoshina, R.; Ogura, A. De novo assembly of middle-sized genome using MinION and Illumina sequencers. BMC Genom. 2018, 19, 700. [Google Scholar] [CrossRef] [PubMed]
- Solares, E.A.; Chakraborty, M.; Miller, D.E.; Kalsow, S.; Hall, K.; Perera, A.G.; Emerson, J.J.; Hawley, R.S. Rapid Low-Cost Assembly of the Drosophila melanogaster Reference Genome Using Low-Coverage, Long-Read Sequencing. G3 Genes Genomes Genet. 2018, 8, 3143–3154. [Google Scholar] [CrossRef] [PubMed]
- Tyson, J.R.; O’Neil, N.J.; Jain, M.; Olsen, H.E.; Hieter, P.; Snutch, T.P. MinION-based long-read sequencing and assembly extends the Caenorhabditis elegans reference genome. Genome Res. 2018, 28, 266–274. [Google Scholar] [CrossRef] [PubMed]
- Greninger, A.L.; Naccache, S.N.; Federman, S.; Yu, G.; Mbala, P.; Bres, V.; Stryke, D.; Bouquet, J.; Somasekar, S.; Linnen, J.M.; et al. Rapid metagenomic identification of viral pathogens in clinical samples by real-time nanopore sequencing analysis. Genome Med. 2015, 7. [Google Scholar] [CrossRef]
- Hoenen, T.; Groseth, A.; Rosenke, K.; Fischer, R.J.; Hoenen, A.; Judson, S.D.; Martellaro, C.; Falzarano, D.; Marzi, A.; Squires, R.B.; et al. Nanopore Sequencing as a Rapidly Deployable Ebola Outbreak Tool. Emerg. Infect. Dis. 2016, 22, 331–334. [Google Scholar] [CrossRef]
- Mataseje, L.F.; Abdesselam, K.; Vachon, J.; Mitchel, R.; Bryce, E.; Roscoe, D.; Boyd, D.A.; Embree, J.; Katz, K.; Kibsey, P.; et al. Results from the Canadian Nosocomial Infection Surveillance Program on Carbapenemase-Producing Enterobacteriaceae, 2010 to 2014. Antimicrob. Agents Chemother. 2016, 60, 6787–6794. [Google Scholar] [CrossRef] [PubMed]
- Loose, M.; Malla, S.; Stout, M. Real-time selective sequencing using nanopore technology. Nat. Methods 2016, 13, 751–754. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.; Koren, S.; Miga, K.H.; Quick, J.; Rand, A.C.; Sasani, T.A.; Tyson, J.R.; Beggs, A.D.; Dilthey, A.T.; Fiddes, I.T.; et al. Nanopore sequencing and assembly of a human genome with ultra-long reads. Nat. Biotechnol. 2018, 36, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Sims, D.; Sudbery, I.; Ilott, N.E.; Heger, A.; Ponting, C.P. Sequencing depth and coverage: key considerations in genomic analyses. Nat. Rev. Genet. 2014, 15, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef] [PubMed]
- Lim, Y.W.; Cuevas, D.A.; Silva, G.G.Z.; Aguinaldo, K.; Dinsdale, E.A.; Haas, A.F.; Hatay, M.; Sanchez, S.E.; Wegley-Kelly, L.; Dutilh, B.E.; et al. Sequencing at sea: challenges and experiences in Ion Torrent PGM sequencing during the 2013 Southern Line Islands Research Expedition. PeerJ 2014, 2, e520. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Johri, S.; Doane, M.P.; Allen, L.; Dinsdale, E.A. Taking Advantage of the Genomics Revolution for Monitoring and Conservation of Chondrichthyan Populations. Diversity 2019, 11, 49. https://doi.org/10.3390/d11040049
Johri S, Doane MP, Allen L, Dinsdale EA. Taking Advantage of the Genomics Revolution for Monitoring and Conservation of Chondrichthyan Populations. Diversity. 2019; 11(4):49. https://doi.org/10.3390/d11040049
Chicago/Turabian StyleJohri, Shaili, Michael P. Doane, Lauren Allen, and Elizabeth A. Dinsdale. 2019. "Taking Advantage of the Genomics Revolution for Monitoring and Conservation of Chondrichthyan Populations" Diversity 11, no. 4: 49. https://doi.org/10.3390/d11040049
APA StyleJohri, S., Doane, M. P., Allen, L., & Dinsdale, E. A. (2019). Taking Advantage of the Genomics Revolution for Monitoring and Conservation of Chondrichthyan Populations. Diversity, 11(4), 49. https://doi.org/10.3390/d11040049