Aquatic Hemiptera in Southwest Cameroon: Biodiversity of Potential Reservoirs of Mycobacterium ulcerans and Multiple Wolbachia Sequence Types Revealed by Metagenomics

 ,
, 
Abstract

1. Introduction
2. Materials and Methods
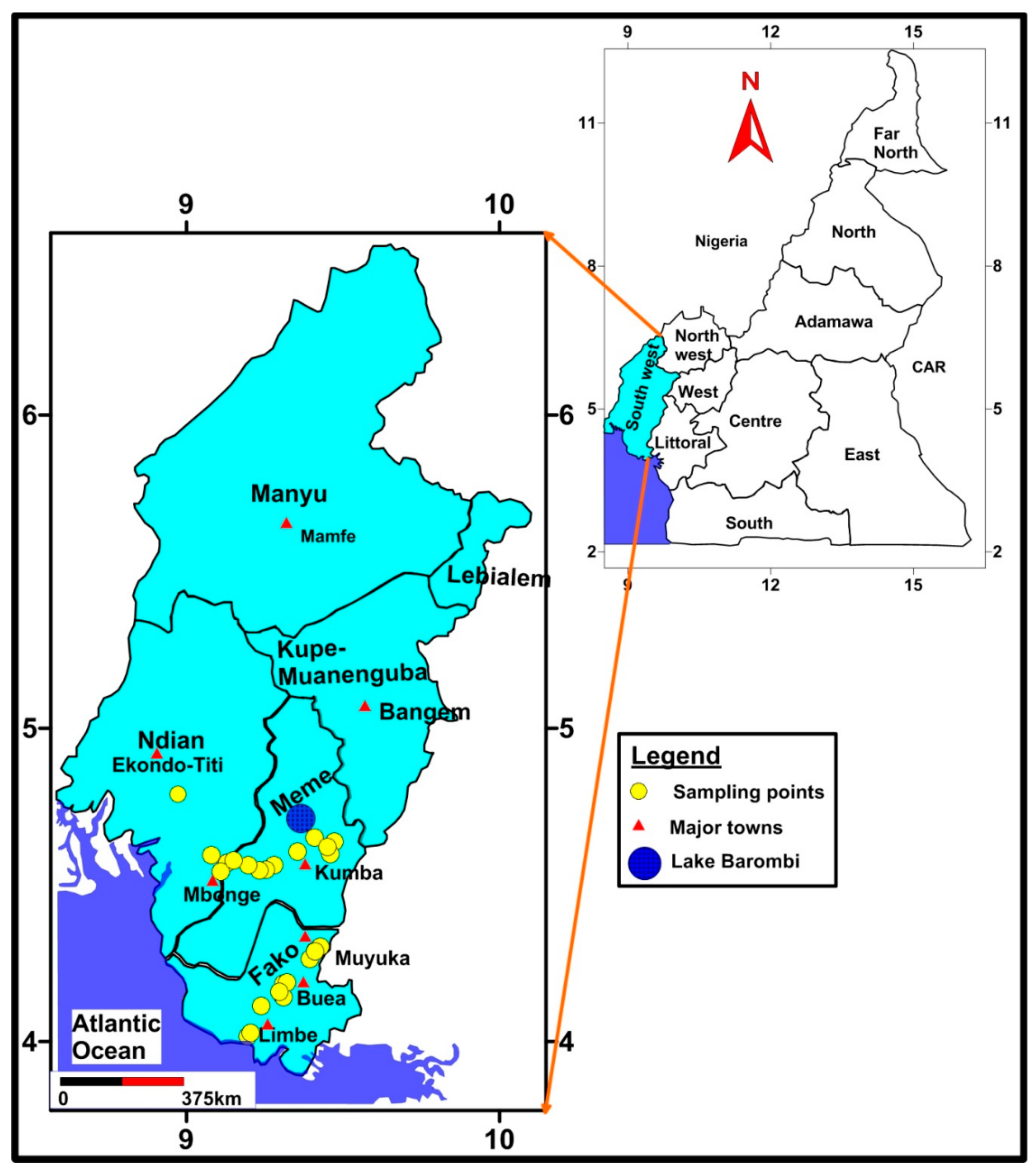
2.1. Study Sites
2.2. Sample Collection and Transportation
2.3. Aquatic Bugs Identification and Morphotyping
2.4. DNA Extraction from Aquatic Bugs
2.5. Coi Amplification (DNA Barcoding)
2.6. Amplification of a Mycobacterial rpoB Gene Fragment by Quantitative PCR
2.7. Confirmatory Conventional PCRs for Bacteria
2.8. Metagenomic Sequencing
2.9. Taxonomic Assignment
2.10. Mitogenome Assembly and Annotation
2.11. Barcode Analysis of coi Sequences and Phylogenetics
2.12. Analysis of Wolbachia Genes and Phylogenetics
3. Results
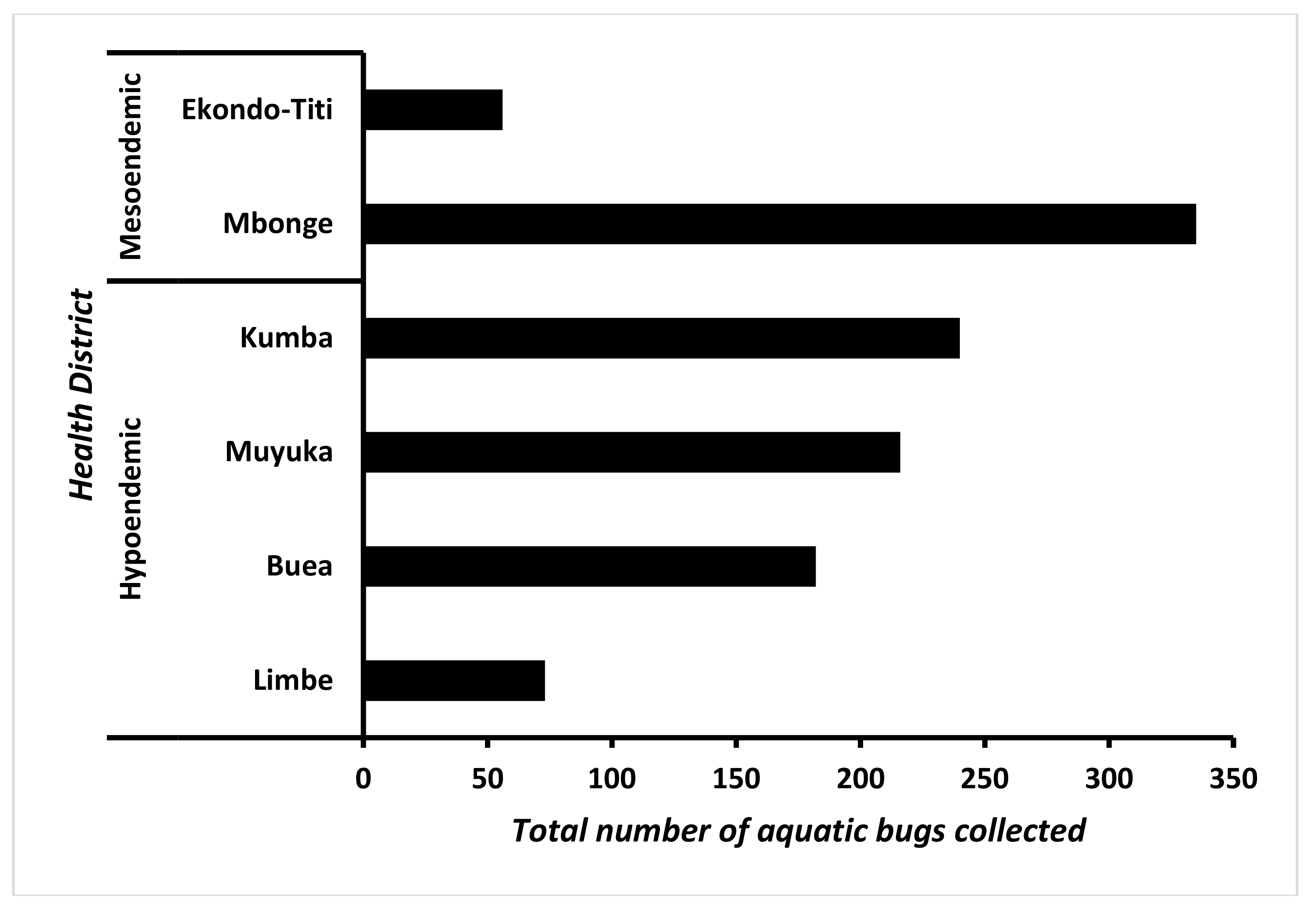
3.1. Distribution of Aquatic Bugs in the Health Districts
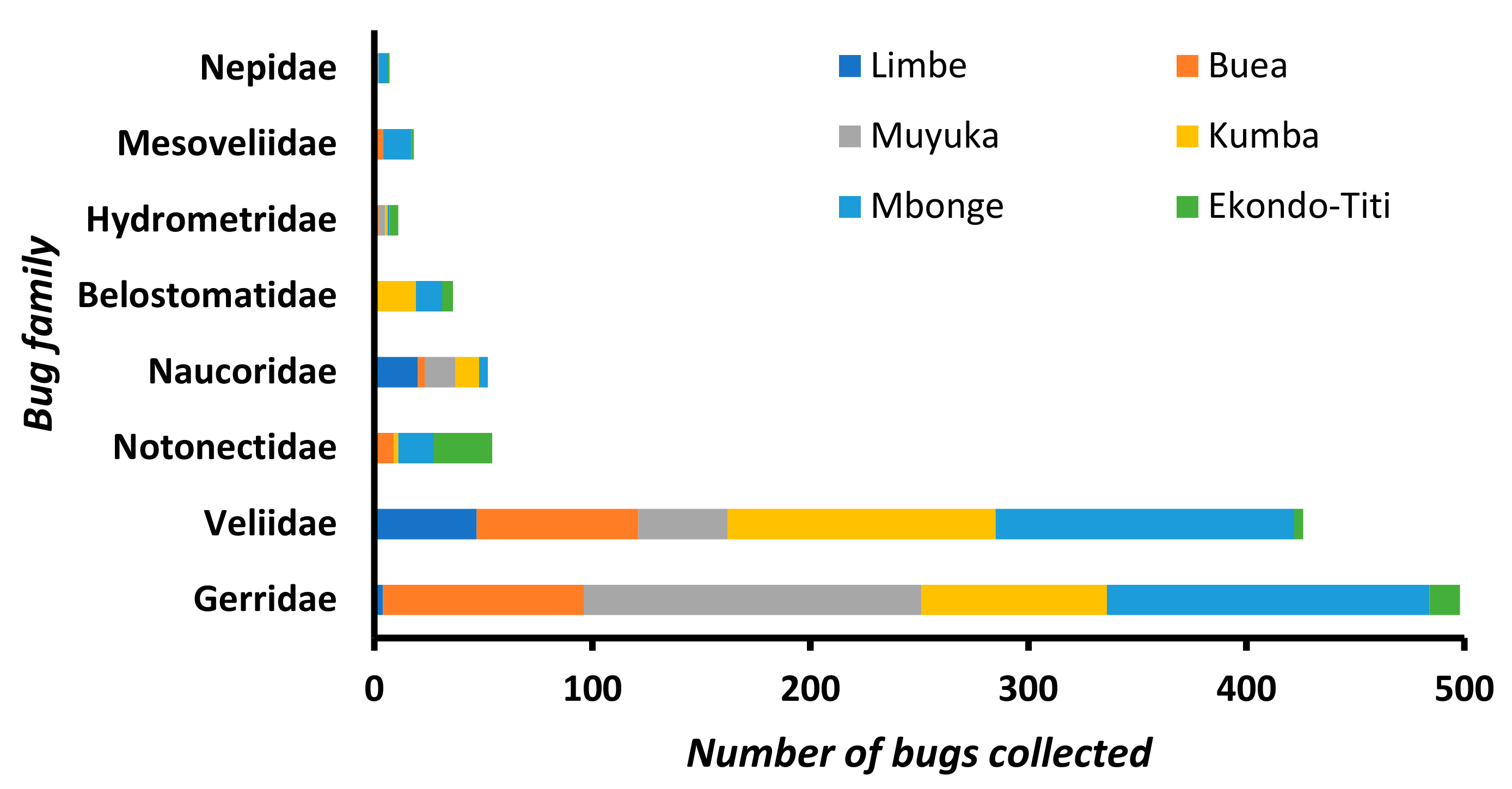
3.2. Composition of Aquatic Bug Taxa
3.3. Prevalence of Mycobacterial DNA in the Aquatic Bugs
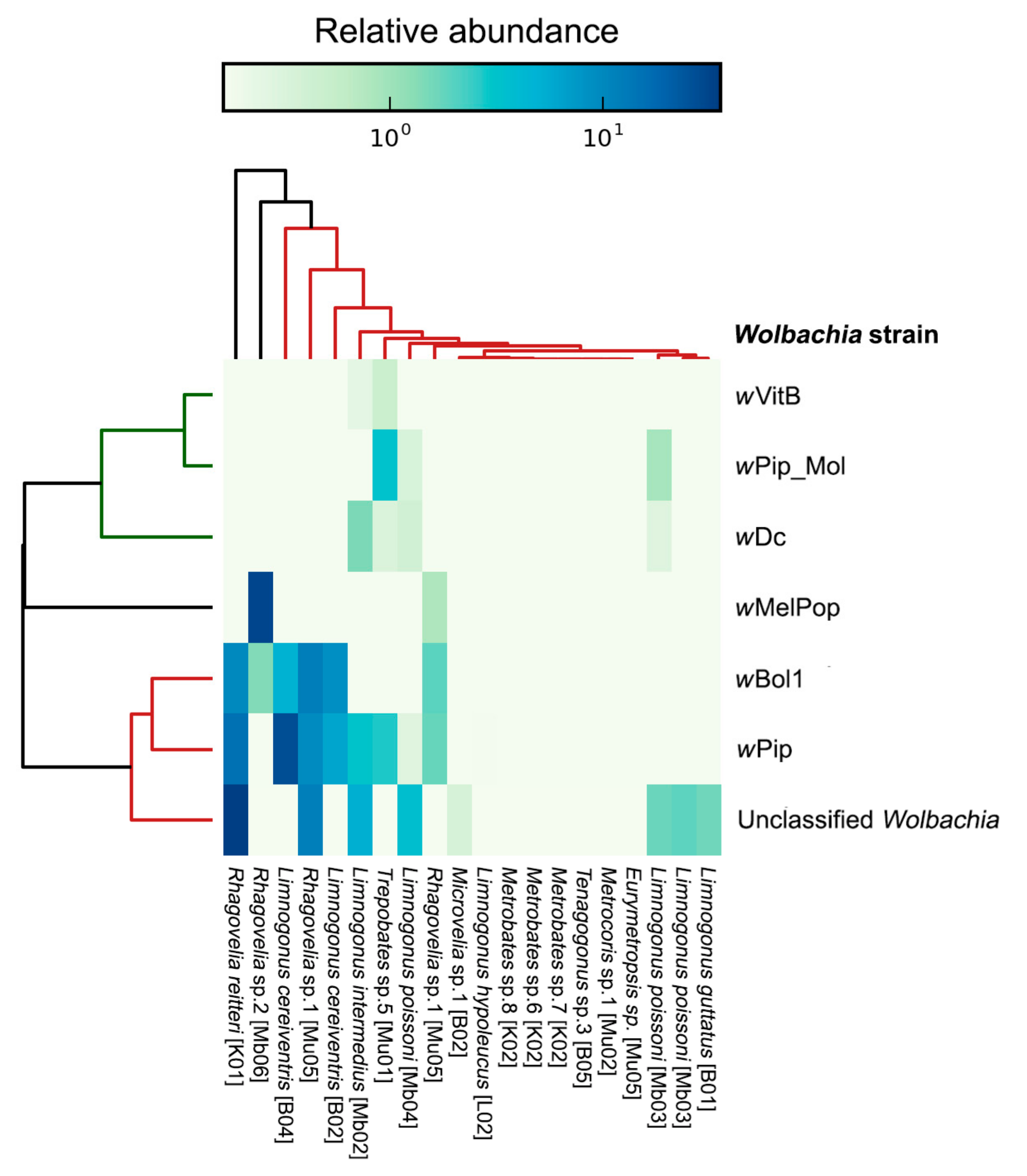
3.4. Bacterial Sequences in Gerrid and Veliid Metagenomic Datasets
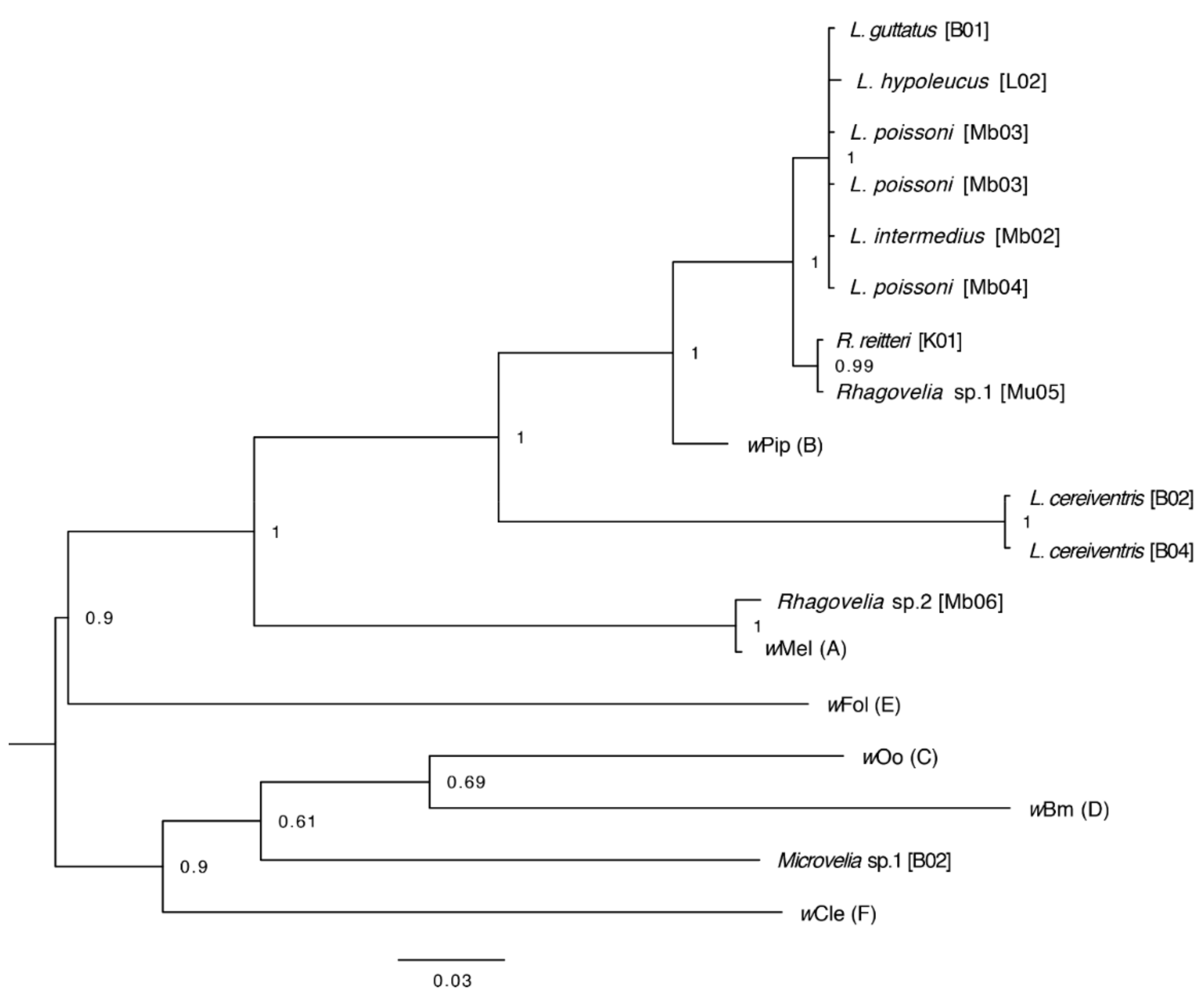
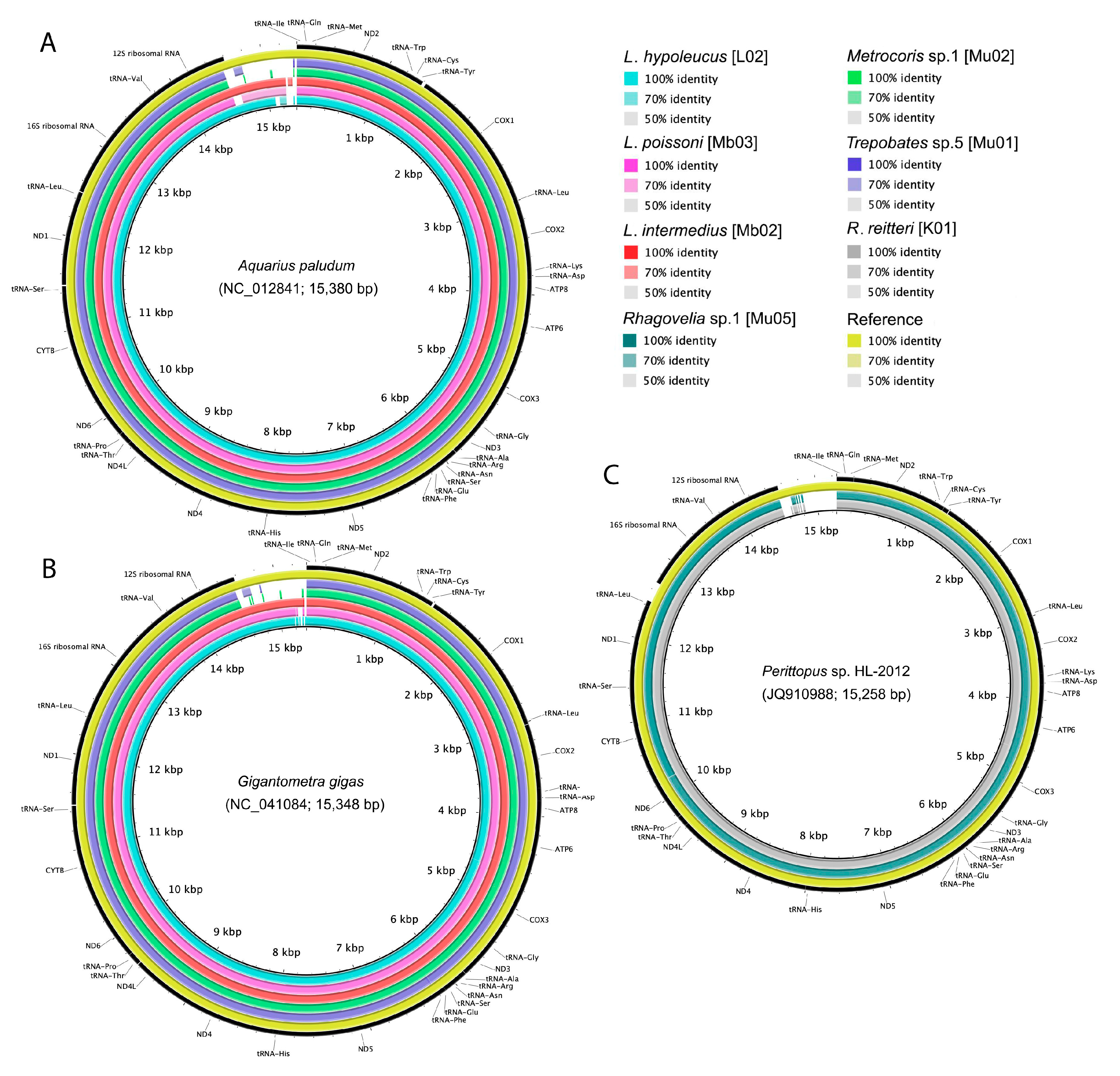
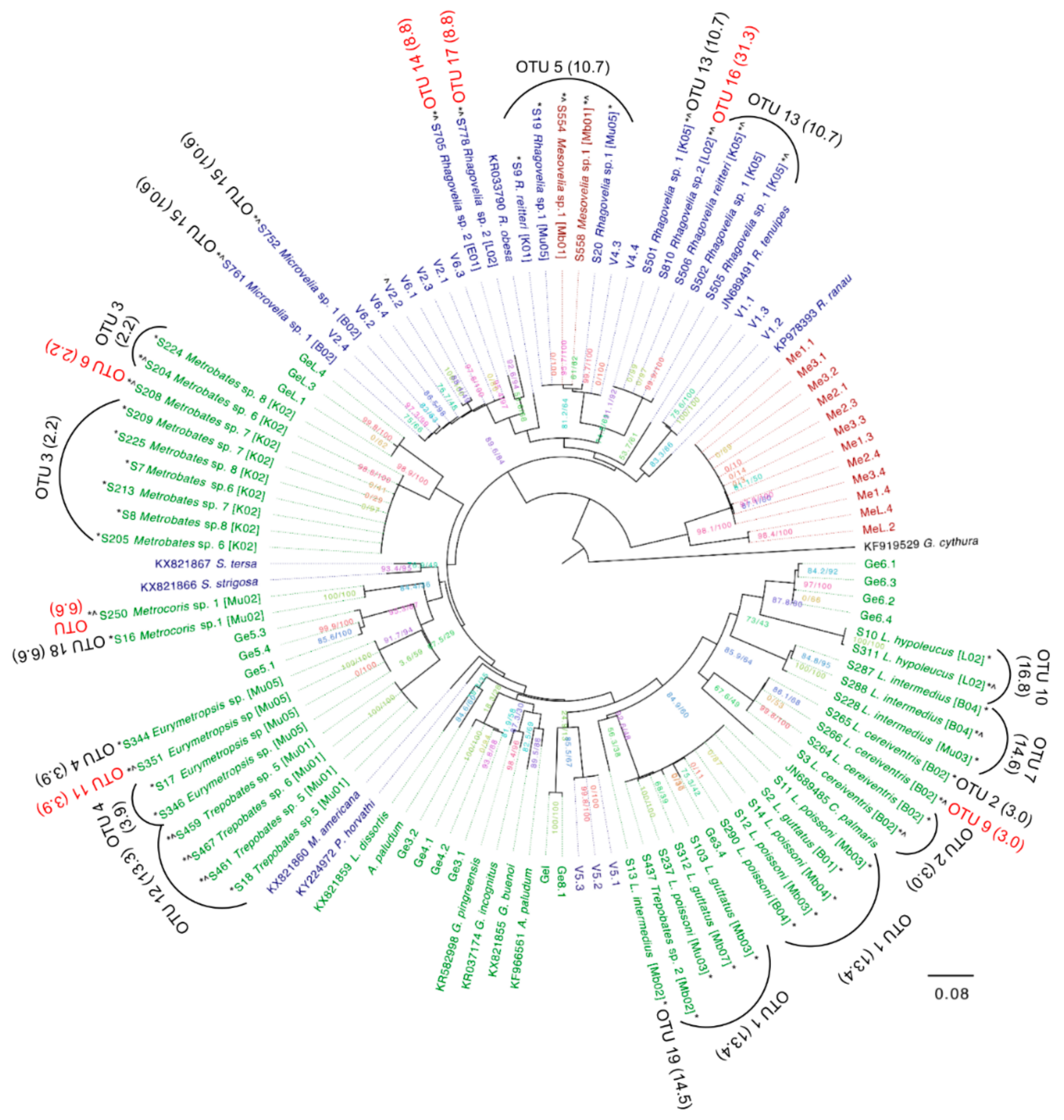
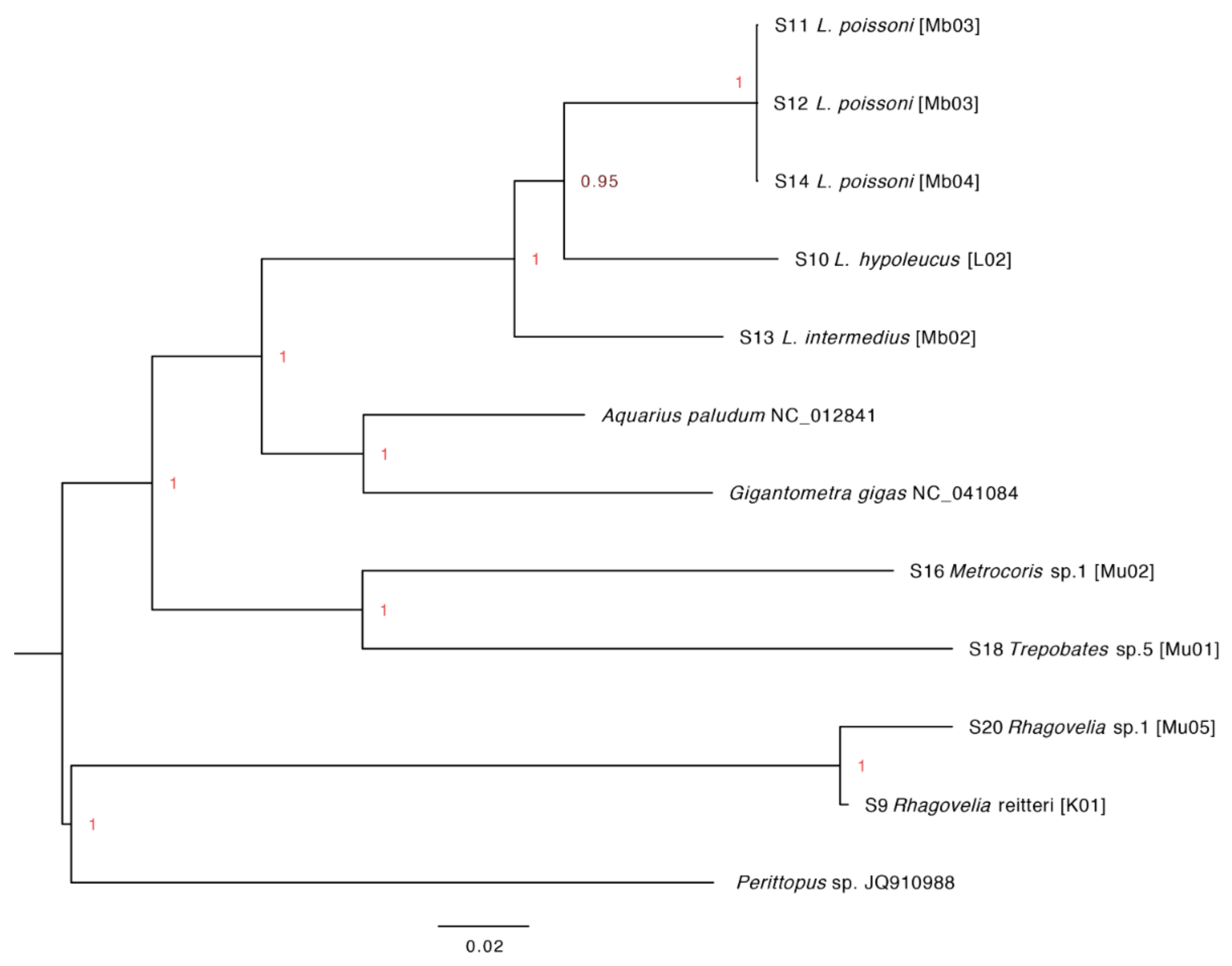
3.5. Assembly of Complete Mitogenomes from Gerrids and Veliids and DNA Barcoding
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Tai, A.Y.C.; Athan, E.; Friedman, N.D.; Hughes, A.; Walton, A.; O’Brien, D.P. Increased severity and spread of Mycobacterium ulcerans, Southeastern Australia. Emerg. Infect. Dis. 2018, 24, 58–64. [Google Scholar] [CrossRef]
- Röltgen, K.; Pluschke, G. Epidemiology and disease burden of Buruli ulcer: A review. Res. Rep. Trop. Med. 2015, 6, 59–73. [Google Scholar]
- Toutous Trellu, L.; Nkemenang, P.; Comte, E.; Ehounou, G.; Atangana, P.; Mboua, D.J.; Rusch, B.; Njih Tabah, E.; Etard, J.F.; Mueller, Y.K. Differential diagnosis of skin ulcers in a Mycobacterium ulcerans endemic area: Data from a prospective study in Cameroon. PLoS Negl. Trop. Dis. 2016, 10, e0004385. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, K.H.; Padgett, J.J. Risk factors for Mycobacterium ulcerans infection. Int. J. Infect. Dis. 2010, 14, e677–e681. [Google Scholar] [CrossRef] [PubMed]
- Loftus, M.J.; Trubiano, J.A.; Tay, E.L.; Lavender, C.J.; Globan, M.; Fyfe, J.A.M.; Johnson, P.D.R. The incubation period of Buruli ulcer (Mycobacterium ulcerans infection) in Victoria, Australia remains similar despite changing geographic distribution of disease. PLoS Negl. Trop. Dis. 2018, 12, e0006323. [Google Scholar] [CrossRef] [PubMed]
- Trubiano, J.A.; Lavender, C.J.; Fyfe, J.A.; Bittmann, S.; Johnson, P.D. The incubation period of Buruli ulcer (Mycobacterium ulcerans infection). PLoS Negl. Trop. Dis. 2013, 7, e2463. [Google Scholar] [CrossRef] [PubMed]
- Doig, K.D.; Holt, K.E.; Fyfe, J.A.; Lavender, C.J.; Eddyani, M.; Portaels, F.; Yeboah-Manu, D.; Pluschke, G.; Seemann, T.; Stinear, T.P. On the origin of Mycobacterium ulcerans, the causative agent of Buruli ulcer. BMC Genom. 2012, 13, 258. [Google Scholar] [CrossRef] [PubMed]
- George, K.M.; Chatterjee, D.; Gunawardana, G.; Welty, D.; Hayman, J.; Lee, R.; Small, P.L. Mycolactone: A polyketide toxin from Mycobacterium ulcerans required for virulence. Science 1999, 283, 854–857. [Google Scholar] [CrossRef]
- Fraga, A.G.; Cruz, A.; Martins, T.G.; Torrado, E.; Saraiva, M.; Pereira, D.R.; Meyers, W.M.; Portaels, F.; Silva, M.T.; Castro, A.G.; et al. Mycobacterium ulcerans triggers T-cell immunity followed by local and regional but not systemic immunosuppression. Infect. Immun. 2011, 79, 421–430. [Google Scholar] [CrossRef]
- Simpson, H.; Deribe, K.; Tabah, E.N.; Peters, A.; Maman, I.; Frimpong, M.; Ampadu, E.; Phillips, R.; Saunderson, P.; Pullan, R.L.; et al. Mapping the global distribution of Buruli ulcer: A systematic review with evidence consensus. Lancet Glob. Health 2019, 7, e912–e922. [Google Scholar] [CrossRef]
- Pouillot, R.; Matias, G.; Wondje, C.M.; Portaels, F.; Valin, N.; Ngos, F.; Njikap, A.; Marsollier, L.; Fontanet, A.; Eyangoh, S. Risk factors for Buruli ulcer: A case control study in Cameroon. PLoS Negl. Trop. Dis. 2007, 1, e101. [Google Scholar] [CrossRef] [PubMed]
- Ravisse, P. Skin ulcer caused by Mycobacterium ulcerans in Cameroon. I. Clinical, epidemiological and histological study. Bull. Soc. Pathol. Exot. 1977, 70, 109–124. [Google Scholar]
- MacCallum, P.; Tolhurst, J.C.; Buckle, G.; Sissons, H.A. A new mycobacterial infection in man. J. Pathol. Bacteriol. 1948, 60, 93–122. [Google Scholar] [CrossRef] [PubMed]
- Dodge, O.G.; Lunn, H.F. Buruli ulcer: A mycobacterial skin ulcer in a Uganda child. J. Trop. Med. Hyg. 1962, 65, 139–142. [Google Scholar]
- Tabah, E.N.; Nsagha, D.S.; Bissek, A.C.; Njamnshi, A.K.; Bratschi, M.W.; Pluschke, G.; Um Boock, A. Buruli ulcer in Cameroon: The development and impact of the national control programme. PLoS Negl. Trop. Dis. 2016, 10, e0004224. [Google Scholar] [CrossRef]
- Akoachere, J.F.; Nsai, F.S.; Ndip, R.N. A community based study on the mode of transmission, prevention and treatment of Buruli ulcers in Southwest Cameroon: Knowledge, attitude and practices. PLoS ONE 2016, 11, e0156463. [Google Scholar] [CrossRef]
- Marion, E.; Landier, J.; Boisier, P.; Marsollier, L.; Fontanet, A.; Le Gall, P.; Aubry, J.; Djeunga, N.; Umboock, A.; Eyangoh, S. Geographic expansion of Buruli ulcer disease, Cameroon. Emerg. Infect. Dis. 2011, 17, 551–553. [Google Scholar] [CrossRef]
- Noeske, J.; Kuaban, C.; Rondini, S.; Sorlin, P.; Ciaffi, L.; Mbuagbaw, J.; Portaels, F.; Pluschke, G. Buruli ulcer disease in Cameroon rediscovered. Am. J. Trop. Med. Hyg. 2004, 70, 520–526. [Google Scholar] [CrossRef]
- Marion, E.; Eyangoh, S.; Yeramian, E.; Doannio, J.; Landier, J.; Aubry, J.; Fontanet, A.; Rogier, C.; Cassisa, V.; Cottin, J.; et al. Seasonal and regional dynamics of M. ulcerans transmission in environmental context: Deciphering the role of water bugs as hosts and vectors. PLoS Negl. Trop. Dis. 2010, 4, e731. [Google Scholar] [CrossRef]
- Portaels, F.; Meyers, W.M.; Ablordey, A.; Castro, A.G.; Chemlal, K.; de Rijk, P.; Elsen, P.; Fissette, K.; Fraga, A.G.; Lee, R.; et al. First cultivation and characterization of Mycobacterium ulcerans from the environment. PLoS Negl. Trop. Dis. 2008, 2, e178. [Google Scholar] [CrossRef]
- Marsollier, L.; Stinear, T.; Aubry, J.; Saint Andre, J.P.; Robert, R.; Legras, P.; Manceau, A.L.; Audrain, C.; Bourdon, S.; Kouakou, H.; et al. Aquatic plants stimulate the growth of and biofilm formation by Mycobacterium ulcerans in axenic culture and harbor these bacteria in the environment. Appl. Environ. Microbiol. 2004, 70, 1097–1103. [Google Scholar] [CrossRef] [PubMed]
- Willson, S.J.; Kaufman, M.G.; Merritt, R.W.; Williamson, H.R.; Malakauskas, D.M.; Benbow, M.E. Fish and amphibians as potential reservoirs of Mycobacterium ulcerans, the causative agent of Buruli ulcer disease. Infect. Ecol. Epidemiol. 2013, 3, 19946. [Google Scholar]
- Johnson, P.D.; Azuolas, J.; Lavender, C.J.; Wishart, E.; Stinear, T.P.; Hayman, J.A.; Brown, L.; Jenkin, G.A.; Fyfe, J.A. Mycobacterium ulcerans in mosquitoes captured during outbreak of Buruli ulcer, Southeastern Australia. Emerg. Infect. Dis. 2007, 13, 1653–1660. [Google Scholar] [CrossRef] [PubMed]
- Ross, B.C.; Johnson, P.D.; Oppedisano, F.; Marino, L.; Sievers, A.; Stinear, T.; Hayman, J.A.; Veitch, M.G.; Robins-Browne, R.M. Detection of Mycobacterium ulcerans in environmental samples during an outbreak of ulcerative disease. Appl. Environ. Microbiol. 1997, 63, 4135–4138. [Google Scholar]
- Tian, R.B.; Niamke, S.; Tissot-Dupont, H.; Drancourt, M. Detection of Mycobacterium ulcerans DNA in the environment, Ivory Coast. PLoS ONE 2016, 11, e0151567. [Google Scholar] [CrossRef]
- Garchitorena, A.; Ngonghala, C.N.; Texier, G.; Landier, J.; Eyangoh, S.; Bonds, M.H.; Guegan, J.F.; Roche, B. Environmental transmission of Mycobacterium ulcerans drives dynamics of Buruli ulcer in endemic regions of Cameroon. Sci. Rep. 2015, 5, 18055. [Google Scholar] [CrossRef]
- Ohtsuka, M.; Kikuchi, N.; Yamamoto, T.; Suzutani, T.; Nakanaga, K.; Suzuki, K.; Ishii, N. Buruli ulcer caused by Mycobacterium ulcerans subsp shinshuense: A rare case of familial concurrent occurrence and detection of insertion sequence 2404 in Japan. JAMA Dermatol. 2014, 150, 64–67. [Google Scholar] [CrossRef]
- Garchitorena, A.; Guegan, J.F.; Leger, L.; Eyangoh, S.; Marsollier, L.; Roche, B. Mycobacterium ulcerans dynamics in aquatic ecosystems are driven by a complex interplay of abiotic and biotic factors. Elife 2015, 4, e07616. [Google Scholar] [CrossRef]
- Portaels, F.; Elsen, P.; Guimaraes-Peres, A.; Fonteyne, P.A.; Meyers, W.M. Insects in the transmission of Mycobacterium ulcerans infection. Lancet 1999, 353, 986. [Google Scholar] [CrossRef]
- Mosi, L.; Williamson, H.; Wallace, J.R.; Merritt, R.W.; Small, P.L. Persistent association of Mycobacterium ulcerans with West African predaceous insects of the family Belostomatidae. Appl. Environ. Microbiol. 2008, 74, 7036–7042. [Google Scholar] [CrossRef]
- Marsollier, L.; Robert, R.; Aubry, J.; Saint Andre, J.P.; Kouakou, H.; Legras, P.; Manceau, A.L.; Mahaza, C.; Carbonnelle, B. Aquatic insects as a vector for Mycobacterium ulcerans. Appl. Environ. Microbiol. 2002, 68, 4623–4628. [Google Scholar] [CrossRef] [PubMed]
- Marion, E.; Chauty, A.; Yeramian, E.; Babonneau, J.; Kempf, M.; Marsollier, L. A case of guilt by association: Water bug bite incriminated in M. ulcerans infection. Int. J. Mycobacteriol. 2014, 3, 158–161. [Google Scholar] [CrossRef] [PubMed]
- Benbow, M.E.; Williamson, H.; Kimbirauskas, R.; McIntosh, M.D.; Kolar, R.; Quaye, C.; Akpabey, F.; Boakye, D.; Small, P.; Merritt, R.W. Aquatic invertebrates as unlikely vectors of Buruli ulcer disease. Emerg. Infect. Dis. 2008, 14, 1247–1254. [Google Scholar] [CrossRef]
- Ebong, S.M.A.; Garcia-Pena, G.E.; Pluot-Sigwalt, D.; Marsollier, L.; Le Gall, P.; Eyangoh, S.; Guegan, J.F. Ecology and feeding habits drive infection of water bugs with Mycobacterium ulcerans. Ecohealth 2017, 14, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.; Duwal, R.K.; Lee, S. COI barcoding of true bugs (Insecta, Heteroptera). Mol. Ecol. Resour. 2011, 11, 266–270. [Google Scholar] [CrossRef] [PubMed]
- Park, D.S.; Foottit, R.; Maw, E.; Hebert, P.D.N. Barcoding bugs: DNA-based identification of the true bugs (Insecta: Hemiptera: Heteroptera). PLoS ONE 2011, 6, e18749. [Google Scholar] [CrossRef]
- Ebong, S.M.A.; Petit, E.; Le Gall, P.; Chen, P.P.; Nieser, N.; Guilbert, E.; Njiokou, F.; Marsollier, L.; Guegan, J.F.; Pluot-Sigwalt, D.; et al. Molecular species delimitation and morphology of aquatic and sub-aquatic bugs (Heteroptera) in Cameroon. PLoS ONE 2016, 11, e0154905. [Google Scholar]
- Sazama, E.J.; Bosch, M.J.; Shouldis, C.S.; Ouellette, S.P.; Wesner, J.S. Incidence of Wolbachia in aquatic insects. Ecol. Evol. 2017, 7, 1165–1169. [Google Scholar] [CrossRef]
- Weinert, L.A.; Araujo-Jnr, E.V.; Ahmed, M.Z.; Welch, J.J. The incidence of bacterial endosymbionts in terrestrial arthropods. Proc. Biol. Sci. 2015, 282, 20150249. [Google Scholar] [CrossRef]
- Teixeira, L.; Ferreira, A.; Ashburner, M. The bacterial symbiont Wolbachia induces resistance to RNA viral infections in Drosophila melanogaster. PLoS Biol. 2008, 6, e2. [Google Scholar] [CrossRef]
- Zele, F.; Nicot, A.; Berthomieu, A.; Weill, M.; Duron, O.; Rivero, A. Wolbachia increases susceptibility to Plasmodium infection in a natural system. Proc. Biol. Sci. 2014, 281, 20132837. [Google Scholar] [CrossRef] [PubMed]
- Graham, R.I.; Grzywacz, D.; Mushobozi, W.L.; Wilson, K. Wolbachia in a major African crop pest increases susceptibility to viral disease rather than protects. Ecol. Lett. 2012, 15, 993–1000. [Google Scholar] [CrossRef] [PubMed]
- Kamtchum-Tatuene, J.; Makepeace, B.L.; Benjamin, L.; Baylis, M.; Solomon, T. The potential role of Wolbachia in controlling the transmission of emerging human arboviral infections. Curr. Opin. Infect. Dis. 2017, 30, 108–116. [Google Scholar] [CrossRef] [PubMed]
- Jeffries, C.L.; Lawrence, G.G.; Golovko, G.; Kristan, M.; Orsborne, J.; Spence, K.; Hurn, E.; Bandibabone, J.; Tantely, L.M.; Raharimalala, F.N.; et al. Novel Wolbachia strains in Anopheles malaria vectors from sub-Saharan Africa. Wellcome Open Res. 2018, 3, 113. [Google Scholar] [CrossRef]
- Fromont, C.; Adair, K.L.; Douglas, A.E. Correlation and causation between the microbiome, Wolbachia and host functional traits in natural populations of drosophilid flies. Mol. Ecol. 2019, 28, 1826–1841. [Google Scholar] [CrossRef]
- Cheng, D.; Chen, S.; Huang, Y.; Pierce, N.E.; Riegler, M.; Yang, F.; Zeng, L.; Lu, Y.; Liang, G.; Xu, Y. Symbiotic microbiota may reflect host adaptation by resident to invasive ant species. PLoS Pathog. 2019, 15, e1007942. [Google Scholar] [CrossRef]
- Bolz, M.; Bratschi, M.W.; Kerber, S.; Minyem, J.C.; Um Boock, A.; Vogel, M.; Bayi, P.F.; Junghanss, T.; Brites, D.; Harris, S.R.; et al. Locally confined clonal complexes of Mycobacterium ulcerans in two Buruli ulcer endemic regions of Cameroon. PLoS Negl. Trop. Dis. 2015, 9, e0003802. [Google Scholar] [CrossRef]
- Damgaard, J.; Buzzetti, F.M.; Mazzucconi, S.A.; Weir, T.A.; Zettel, H. A molecular phylogeny of the pan-tropical pond skater genus Limnogonus Stål 1868 (Hemiptera-Heteroptera: Gerromorpha-Gerridae). Mol. Phylogenet. Evol. 2010, 57, 669–677. [Google Scholar] [CrossRef]
- Tchakonté, S.; Ajeagah, G.A.; Tchatcho, N.L.N.; Camara, A.I.; Diomandé, D.; Ngassam, P. Stream’s water quality and description of some aquatic species of Coleoptera and Hemiptera (Insecta) in Littoral Region of Cameroon. Biodivers. J. 2015, 6, 27–40. [Google Scholar]
- Mbogho, A.Y.; Sites, R.W. Naucoridae Leach, 1815 (Hemiptera: Heteroptera) of Tanzania. Afr. Invertebr. 2013, 54, 513–542. [Google Scholar] [CrossRef]
- Ebong, S.M.; Eyangoh, S.; Marion, E.; Landier, J.; Marsollier, L.; Guegan, J.F.; Legall, P. Survey of water bugs in Bankim, a new Buruli ulcer endemic area in Cameroon. J. Trop. Med. 2012, 2012, 123843. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.P.; Nieser, N.; Zettel, H. The Aquatic and Semiaquatic Bugs (Heteroptera—Nepomorpha & Gerromorpha) of Malesia; Brill: Leiden, The Netherlands, 2005. [Google Scholar]
- Andersen, N.M.; Weir, T.A. Australian Water Bugs. Their Biology and Identification (Hemiptera-Heteroptera, Gerromorpha & Nepomorpha); Apollo Books; CSIRO Publishing: Clayton, Australia, 2004; Volume 14. [Google Scholar]
- Andersen, N.M. The Semiaquatic Bugs (Hemiptera, Gerromorpha). Phylogeny, Adaptations, Biogeography, and Classification; Scandinavian Science Press Ltd.: Ganløse, Denmark, 1982; Volume 3. [Google Scholar]
- De Moor, I.J.; Day, J.A.; De Moor, F.C. Insecta II. Hemiptera, Megaloptera, Neuroptera, Trichoptera and Lepidoptera; Water Research Commission: Pretoria, South Africa, 2003; Volume 8. [Google Scholar]
- Chen, P.P.; Zettel, H. Five new species of the Halobatinae genus Metrocoris Mayr, 1865 (Insecta: Hemiptera: Gerridae) from continental Asia. Ann. Naturhist. Mus. Wien Serie B 1999, 101, 13–32. [Google Scholar]
- Hecher, C.; Zettel, H. Faunistical and morphological notes on Limnogonus subgenus Limnogonoides Poisson 1965 (Heteroptera: Gerridae). Linz. Biol. Beitr. 1996, 28, 325–333. [Google Scholar]
- Schuh, R.T.; Slater, J.A. True Bugs of the World (Hemiptera: Heteroptera) Classification and Natural History; Cornell University Press: Ithaca, NY, USA, 1995. [Google Scholar]
- Dethier, M. Introduction à la morphologie, la biologie et la classification des Hétéroptères. Bull. Romand Entomol. 1981, 1, 11–16. [Google Scholar]
- Poisson, R.A. Contribution à l’étude des Hydrocorises de Madagascar (mission J. Millot et R. Paulian 1949). Mém. Inst. Sci. Madag. 1952, 1, 24–70. [Google Scholar]
- Poisson, R.A. Contribution à la faune du Cameroun Hémiptères aquatiques. Faune Colon. Fr. 1929, 3, 135–164. [Google Scholar]
- Poisson, R.A. De voyage ML. Chopard en Côte d’Ivoire (1938–39)—Hémiptères aquatiques. Rev. Fr. Entomol. 1941, 8, 77–82. [Google Scholar]
- Poisson, R.A. Hydrocorises du Cameroun. Mission, J. Carayon 1947. Rev. Fr. Entomol. 1948, 3, 167–177. [Google Scholar]
- Poisson, R.A.; de Voyage, M.P.P. Grasse en Afrique Occidentale Française. Hémiptères aquatiques. Ann. Soc. Entomol. Fr. 1937, 106, 115–132. [Google Scholar]
- Poisson, R.A. Quelques Hydrocorises nouveaux de l’Afrique du Sud (mission Suédoise Brink et Rüdebeck). Bull. Soc. Sci. Bretagne 1955, 30, 129–140. [Google Scholar]
- Poisson, R.A. Contribution a l’etude des Hydrocorises de Madagascar (Heteroptera). 4e memoire. Mém. Inst. Sci. Madag. 1956, 7, 243–265. [Google Scholar]
- Poisson, R.A. Catalogue des Hétéroptères Hydrocorises Africano-Malgaches de la famille des Nepidae (Ltreille) 1802. Bull. IFAN Tome 1965, 27, 229–269. [Google Scholar]
- Poisson, R.A. Catalogue des insectes Hétéroptères Gerridae Leach, 1807 Africano-Malgaches. Bull. IFAN Tome 1965, 27, 1466–1503. [Google Scholar]
- Poisson, R.A. Catalogue des insectes Hétéroptères Notonectidae Leach, 1815 Africano-Malgaches. Bull. IFAN Tome 1966, 28, 729–768. [Google Scholar]
- Ammazzalorso, A.D.; Zolnik, C.P.; Daniels, T.J.; Kolokotronis, S.O. To beat or not to beat a tick: Comparison of DNA extraction methods for ticks (Ixodes scapularis). PeerJ 2015, 3, e1147. [Google Scholar] [CrossRef] [PubMed]
- Folmer, O.; Black, M.; Hoeh, W.; Lutz, R.; Vrijenhoek, R. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol. Mar. Biol. Biotechnol. 1994, 3, 294–299. [Google Scholar] [PubMed]
- Ablordey, A.; Amissah, D.A.; Aboagye, I.F.; Hatano, B.; Yamazaki, T.; Sata, T.; Ishikawa, K.; Katano, H. Detection of Mycobacterium ulcerans by the loop mediated isothermal amplification method. PLoS Negl. Trop. Dis. 2012, 6, e1590. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Rousset, F.; O’Neil, S. Phylogeny and PCR-based classification of Wolbachia strains using wsp gene sequences. Proc. Biol. Sci. 1998, 265, 509–515. [Google Scholar] [CrossRef]
- Dong, X.F.; Armstrong, S.D.; Xia, D.; Makepeace, B.L.; Darby, A.C.; Kadowaki, T. Draft genome of the honey bee ectoparasitic mite, Tropilaelaps mercedesae, is shaped by the parasitic life history. Gigascience 2017, 6, gix008. [Google Scholar] [CrossRef]
- Wood, D.E.; Salzberg, S.L. Kraken: Ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 2014, 15, R46. [Google Scholar] [CrossRef]
- Ounit, R.; Lonardi, S. Higher classification sensitivity of short metagenomic reads with CLARK-S. Bioinformatics 2016, 32, 3823–3825. [Google Scholar] [CrossRef]
- Ondov, B.D.; Bergman, N.H.; Phillippy, A.M. Interactive metagenomic visualization in a web browser. BMC Bioinform. 2011, 12, 385. [Google Scholar] [CrossRef] [PubMed]
- Truong, D.T.; Franzosa, E.A.; Tickle, T.L.; Scholz, M.; Weingart, G.; Pasolli, E.; Tett, A.; Huttenhower, C.; Segata, N. Metaphlan2 for enhanced metagenomic taxonomic profiling. Nat. Methods 2015, 12, 902–903. [Google Scholar] [CrossRef] [PubMed]
- Zerbino, D.R.; Birney, E. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008, 18, 821–829. [Google Scholar] [CrossRef] [PubMed]
- Laetsch, D.R.; Blaxter, M.L. Blobtools: Interrogation of genome assemblies. F1000Research 2017, 6, 1287. [Google Scholar] [CrossRef]
- Dierckxsens, N.; Mardulyn, P.; Smits, G. Novoplasty: De novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 2017, 45, e18. [Google Scholar]
- Bernt, M.; Donath, A.; Juhling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Putz, J.; Middendorf, M.; Stadler, P.F. Mitos: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef]
- Lowe, T.M.; Chan, P.P. tRNAscan-SE on-line: Integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 2016, 44, W54–W57. [Google Scholar] [CrossRef]
- Alikhan, N.F.; Petty, N.K.; Ben Zakour, N.L.; Beatson, S.A. Blast ring image generator (BRIG): Simple prokaryote genome comparisons. BMC Genom. 2011, 12, 402. [Google Scholar] [CrossRef]
- Li, H.; Leavengood, J.M., Jr.; Chapman, E.G.; Burkhardt, D.; Song, F.; Jiang, P.; Liu, J.; Zhou, X.; Cai, W. Mitochondrial phylogenomics of Hemiptera reveals adaptive innovations driving the diversification of true bugs. Proc. Biol. Sci. 2017, 284, 20171223. [Google Scholar] [CrossRef]
- Hua, J.; Li, M.; Dong, P.; Cui, Y.; Xie, Q.; Bu, W. Phylogenetic analysis of the true water bugs (Insecta: Hemiptera: Heteroptera: Nepomorpha): Evidence from mitochondrial genomes. BMC Evol. Biol. 2009, 9, 134. [Google Scholar] [CrossRef]
- Sun, X.; Wang, Y.; Chen, P.P.; Wang, H.; Lixiang, L.; Ye, Z.; Wu, Y.; Li, T.; Bu, W.; Xie, Q. Biased heteroplasmy within the mitogenomic sequences of Gigantometra gigas revealed by Sanger and high-throughput methods. Zool. Syst. 2018, 43, 356–386. [Google Scholar]
- Darling, A.C.; Mau, B.; Blattner, F.R.; Perna, N.T. Mauve: Multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004, 14, 1394–1403. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Hohna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. Mrbayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Chaisiri, K.; Xia, D.; Armstrong, S.D.; Fang, Y.; Donnelly, M.J.; Kadowaki, T.; McGarry, J.W.; Darby, A.C.; Makepeace, B.L. Genomes of trombidid mites reveal novel predicted allergens and laterally transferred genes associated with secondary metabolism. Gigascience 2018, 7, giy127. [Google Scholar] [CrossRef]
- Huang, X.; Madan, A. Cap3: A DNA sequence assembly program. Genome Res. 1999, 9, 868–877. [Google Scholar] [CrossRef]
- Ratnasingham, S.; Hebert, P.D. Bold: The Barcode of Life data system (http://www.Barcodinglife.Org). Mol. Ecol. Notes 2007, 7, 355–364. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Talavera, G.; Castresana, J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 2007, 56, 564–577. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. Iq-tree: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. Spades: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef] [PubMed]
- Emms, D.M.; Kelly, S. Orthofinder: Solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 2015, 16, 157. [Google Scholar] [CrossRef] [PubMed]
- Bordenstein, S.R.; Bordenstein, S.R. Eukaryotic association module in phage WO genomes from Wolbachia. Nat. Commun. 2016, 7, 13155. [Google Scholar] [CrossRef] [PubMed]
- Gu, H.; Chen, Y.; Liu, X.; Wang, H.; Shen-Tu, J.; Wu, L.; Zeng, L.; Xu, J. The effective migration of Massilia sp. Wf1 by Phanerochaete chrysosporium and its phenanthrene biodegradation in soil. Sci. Total Environ. 2017, 593, 695–703. [Google Scholar] [CrossRef] [PubMed]
- Baldo, L.; Werren, J.H. Revisiting Wolbachia supergroup typing based on WSP: Spurious lineages and discordance with MLST. Curr. Microbiol. 2007, 55, 81–87. [Google Scholar] [CrossRef]
- LePage, D.P.; Metcalf, J.A.; Bordenstein, S.R.; On, J.; Perlmutter, J.I.; Shropshire, J.D.; Layton, E.M.; Funkhouser-Jones, L.J.; Beckmann, J.F.; Bordenstein, S.R. Prophage WO genes recapitulate and enhance Wolbachia-induced cytoplasmic incompatibility. Nature 2017, 543, 243–247. [Google Scholar] [CrossRef]
- Bronstein, O.; Kroh, A.; Haring, E. Mind the gap! The mitochondrial control region and its power as a phylogenetic marker in echinoids. BMC Evol. Biol. 2018, 18, 80. [Google Scholar] [CrossRef]
- Song, H.; Buhay, J.E.; Whiting, M.F.; Crandall, K.A. Many species in one: DNA barcoding overestimates the number of species when nuclear mitochondrial pseudogenes are coamplified. Proc. Natl. Acad. Sci. USA 2008, 105, 13486–13491. [Google Scholar] [CrossRef]
- Garchitorena, A.; Roche, B.; Kamgang, R.; Ossomba, J.; Babonneau, J.; Landier, J.; Fontanet, A.; Flahault, A.; Eyangoh, S.; Guegan, J.F.; et al. Mycobacterium ulcerans ecological dynamics and its association with freshwater ecosystems and aquatic communities: Results from a 12-month environmental survey in Cameroon. PLoS Negl. Trop. Dis. 2014, 8, e2879. [Google Scholar] [CrossRef]
- Hammer, T.J.; Dickerson, J.C.; Fierer, N. Evidence-based recommendations on storing and handling specimens for analyses of insect microbiota. PeerJ 2015, 3, e1190. [Google Scholar] [CrossRef]
- Iturbe-Ormaetxe, I.; Walker, T.; O’Neill, S.L. Wolbachia and the biological control of mosquito-borne disease. EMBO Rep. 2011, 12, 508–518. [Google Scholar] [CrossRef] [PubMed]
- Crainey, J.L.; Wilson, M.D.; Post, R.J. Phylogenetically distinct Wolbachia gene and pseudogene sequences obtained from the African onchocerciasis vector Simulium squamosum. Int. J. Parasitol. 2010, 40, 569–578. [Google Scholar] [CrossRef] [PubMed]
- Woodford, L.; Bianco, G.; Ivanova, Y.; Dale, M.; Elmer, K.; Rae, F.; Larcombe, S.D.; Helm, B.; Ferguson, H.M.; Baldini, F. Vector species-specific association between natural Wolbachia infections and avian malaria in black fly populations. Sci. Rep. 2018, 8, 4188. [Google Scholar] [CrossRef] [PubMed]
- Cordaux, R.; Pichon, S.; Hatira, H.B.; Doublet, V.; Greve, P.; Marcade, I.; Braquart-Varnier, C.; Souty-Grosset, C.; Charfi-Cheilchrouha, F.; Bouchon, D. Widespread Wolbachia infection in terrestrial isopods and other crustaceans. Zookeys 2012, 176, 123–131. [Google Scholar] [CrossRef]
- Baltanas, A.; Zabal-Aguirre, M.; Pita, M.; Lopez-Fernandez, C. Wolbachia identified in a new crustacean host: An explanation of the prevalence of asexual reproduction in non-marine ostracods? Fund. Appl. Limnol. 2007, 169, 217–221. [Google Scholar] [CrossRef]
- Aikawa, T.; Anbutsu, H.; Nikoh, N.; Kikuchi, T.; Shibata, F.; Fukatsu, T. Longicorn beetle that vectors pinewood nematode carries many Wolbachia genes on an autosome. Proc. R. Soc. B Biol. Sci. 2009, 276, 3791–3798. [Google Scholar] [CrossRef]
- McNulty, S.N.; Foster, J.M.; Mitreva, M.; Hotopp, J.C.D.; Martin, J.; Fischer, K.; Wu, B.; Davis, P.J.; Kumar, S.; Brattig, N.W.; et al. Endosymbiont DNA in endobacteria-free filarial nematodes indicates ancient horizontal genetic transfer. PLoS ONE 2010, 5, e11029. [Google Scholar] [CrossRef]
- Koutsovoulos, G.; Makepeace, B.; Tanya, V.N.; Blaxter, M. Palaeosymbiosis revealed by genomic fossils of Wolbachia in a strongyloidean nematode. PLoS Genet. 2014, 10, e1004397. [Google Scholar] [CrossRef]
- Funkhouser-Jones, L.J.; Sehnert, S.R.; Martinez-Rodriguez, P.; Toribio-Fernandez, R.; Pita, M.; Bella, J.L.; Bordenstein, S.R. Wolbachia co-infection in a hybrid zone: Discovery of horizontal gene transfers from two Wolbachia supergroups into an animal genome. PeerJ 2015, 3, e1479. [Google Scholar] [CrossRef]
- LePage, D.; Bordenstein, S.R. Wolbachia: Can we save lives with a great pandemic? Trends Parasitol. 2013, 29, 385–393. [Google Scholar] [CrossRef]
- Gerth, M.; Gansauge, M.T.; Weigert, A.; Bleidorn, C. Phylogenomic analyses uncover origin and spread of the Wolbachia pandemic. Nat. Commun. 2014, 5, 5117. [Google Scholar] [CrossRef] [PubMed]
- Armbruster, P.; Damsky, W.E.; Giordano, R.; Birungi, J.; Munstermann, L.E.; Conn, J.E. Infection of New- and Old-world Aedes albopictus (Diptera: Culicidae) by the intracellular parasite Wolbachia: Implications for host mitochondrial DNA evolution. J. Med. Entomol. 2003, 40, 356–360. [Google Scholar] [CrossRef] [PubMed]
- Ellegaard, K.M.; Klasson, L.; Naslund, K.; Bourtzis, K.; Andersson, S.G.E. Comparative genomics of Wolbachia and the bacterial species concept. PLoS Genet. 2013, 9, e1003381. [Google Scholar] [CrossRef] [PubMed]
- Casiraghi, M.; Bordenstein, S.R.; Baldo, L.; Lo, N.; Beninati, T.; Wernegreen, J.J.; Werren, J.H.; Bandi, C. Phylogeny of Wolbachia pipientis based on glta, groel and ftsz gene sequences: Clustering of arthropod and nematode symbionts in the F supergroup, and evidence for further diversity in the Wolbachia tree. Microbiol. SGM 2005, 151, 4015–4022. [Google Scholar] [CrossRef] [PubMed]
- Hosokawa, T.; Koga, R.; Kikuchi, Y.; Meng, X.Y.; Fukatsu, T. Wolbachia as a bacteriocyte-associated nutritional mutualist. Proc. Natl. Acad. Sci. USA 2010, 107, 769–774. [Google Scholar] [CrossRef] [PubMed]
- Tijsse-Klasen, E.; Braks, M.; Scholte, E.J.; Sprong, H. Parasites of vectors—Ixodiphagus hookeri and its Wolbachia symbionts in ticks in the Netherlands. Parasit. Vectors 2011, 4, 228. [Google Scholar] [CrossRef]
- Chrostek, E.; Gerth, M. Is Anopheles gambiae a natural host of Wolbachia? MBio 2019, 10, e00784-19. [Google Scholar] [CrossRef]
- Cariou, M.; Duret, L.; Charlat, S. The global impact of Wolbachia on mitochondrial diversity and evolution. J. Evol. Biol. 2017, 30, 2204–2210. [Google Scholar] [CrossRef]
- Zabal-Aguirre, M.; Arroyo, F.; Garcia-Hurtado, J.; de la Torre, J.; Hewitt, G.M.; Bella, J.L. Wolbachia effects in natural populations of Chorthippus parallelus from the Pyrenean hybrid zone. J. Evol. Biol. 2014, 27, 1136–1148. [Google Scholar] [CrossRef]
- Raychoudhury, R.; Baldo, L.; Oliveira, D.C.; Werren, J.H. Modes of acquisition of Wolbachia: Horizontal transfer, hybrid introgression, and codivergence in the Nasonia species complex. Evolution 2009, 63, 165–183. [Google Scholar] [CrossRef]
- Miyata, M.; Konagaya, T.; Yukuhiro, K.; Nomura, M.; Kageyama, D. Wolbachia-induced meiotic drive and feminization is associated with an independent occurrence of selective mitochondrial sweep in a butterfly. Biol. Lett. 2017, 13, 20170153. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Family (n) | Genus | Putative Species | Total | No. of Morphotypes |
|---|---|---|---|---|
| Gerridae (498) | Limnogonus | Limnogonus (L.) curriei Bergroth 1916 | 2 | 1 |
| Limnogonus (L.) guttatus Poisson 1948 | 45 | 1 | ||
| Limnogonus (L.) hypoleucus Gerstaecker 1873 | 4 | 1 | ||
| Limnogonus (L.) intermedius Poisson 1941 | 57 | 1 | ||
| Limnogonus (L.) poissoni Andersen 1975 | 83 | 1 | ||
| Limnogonus (s. str.) cereinventris Signoret 1862 | 30 | 1 | ||
| Limnogonus (L.) spp. | 41 | 7 | ||
| Limnogonus (s. str.) sp. | 1 | 1 | ||
| Aquarius | Aquarius remigis | 3 | 1 | |
| Aquarius spp. | 6 | 3 | ||
| Tenagogonus | Tenagogonus olbovitlotus | 6 | 1 | |
| Tenagogonus spp. | 8 | 4 | ||
| Trepobates | Trepobates spp. | 89 | 13 | |
| Metrobates | Metrobates spp. | 55 | 10 | |
| Metrocoris | Metrocoris spp. | 16 | 4 | |
| Neogerris | Neogerris severini | 1 | 1 | |
| Rhagadotarsus | Rhagadotarsus sp. | 1 | 1 | |
| Eurymetra | Eurymetra sp. | 1 | 1 | |
| Eurymetropsis | Eurymetropsis sp. | 50 | 1 | |
| Veliidae (426) | Rhagovelia | Rhagovelia reitteri Reuters, 1882 | 184 | 1 |
| Rhagovelia spp. | 195 | 9 | ||
| Microvelia | Microvelia spp. | 47 | 6 | |
| Notonectidae (54) | Walambianisops | Walambianisops spp. | 20 | 5 |
| Anisops | Anisops spp. | 11 | 3 | |
| Paranisops | Paranisops sp. | 1 | 1 | |
| Enithares | Enithares sp. | 5 | 2 | |
| Notonecta | Notonecta sp. | 2 | 1 | |
| Nychia | Nychia sp. | 15 | 4 | |
| Naucoridae (52) | Aneurocoris | Aneurocoris sp. | 25 | 1 |
| Illyocoris | Illyocoris sp. | 6 | 2 | |
| Laccocoris | Laccocoris sp. | 5 | 1 | |
| Maccrocolis | Maccrocolis laticollis | 2 | 1 | |
| Naucoris | Naucoris obscuratus | 2 | 1 | |
| Naucoris sp. | 9 | 3 | ||
| Neomaccrocoris | Neomaccrocoris parviceps ocellatus | 2 | 1 | |
| Neomaccrocoris parviceps parviceps | 1 | 1 | ||
| Belostomatidae (36) | Belostoma | Belostoma sp. | 1 | 1 |
| Diplonychus | Diplonychus sp. | 34 | 6 | |
| Lethocerus | Lethocerus sp. | 1 | 1 | |
| Mesoveliidae (18) | Mesovelia | Mesovelia sp. | 18 | 3 |
| Hydrometridae (11) | Hydrometra | Hydrometra sp. | 11 | 1 |
| Nepidae (7) | Ranatra | Ranatra sp. | 7 | 1 |
| Total | 1102 | 110 | ||
| Family (n). | Genus | Putative Species | Total Analyzed | rpoB Positive (%) | Mean rpoB Copy no. |
|---|---|---|---|---|---|
| Gerridae (405) | Limnogonus | Limnogonus (L.) guttatus Poisson 1948 | 39 | 7 (17.9) | 63 |
| Limnogonus (L.) poissoni Andersen 1975 | 76 | 15 (19.7) | 143 | ||
| Limnogonus (L.) intermedius Poisson 1941 | 50 | 7 (14) | 82 | ||
| Limnogonus (L.) curriei Bergroth 1916 | 2 | 0 (0.0) | 0 | ||
| Limnogonus (s. str.) cereinventris Signoret 1862 | 24 | 7 (29.2) | 80 | ||
| Limnogonus (L.) hypoleucus Gerstaecker 1873 | 2 | 1 (50) | 58 | ||
| Limnogonus spp. | 34 | 0 (0.0) | 0 | ||
| Aquarius | Aquarius spp. | 5 | 0 (0.0) | 0 | |
| Tenagogonus | Tenagogonus spp. | 8 | 2 (25.0) | 13 | |
| Metrobates | Metrobates spp. | 42 | 5 (11.9) | 47 | |
| Trepobates | Trepobates spp. | 70 | 4 (5.7) | 50 | |
| Eurymetropsis | Eurymetropsis sp. | 46 | 4 (8.7) | 112 | |
| Metrocoris | Metrocoris spp. | 7 | 1 (14.3) | 91 | |
| Veliidae (366) | Rhagovelia | Rhagovelia reitteri Reuters 1882 | 171 | 3 (1.8) | 44 |
| Rhagovelia spp. | 164 | 11 (6.7) | 111 | ||
| Microvelia | Microvelia spp. | 31 | 2 (6.5) | 33 | |
| Total | 771 | 69 (8.9) | |||
| Sample | Health District | Location Code | Species | Total Reads | All Bacterial Read Pairs (% Total) | Mycobacterium Read Pairs (% Bacterial) | M. Ulcerans Read Pairs (% Bacterial) | Wolbachia Read Pairs (% Bacterial) | Wsp PCR |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Buea | B05 | Tenagogonus sp. 3 | 14,767,852 | 58,114 (0.39%) | 82 (0.14%) | 0 (0.00%) | 125 (0.22%) | - |
| 2 | Buea | B01 | Limnogonus guttatus | 13,155,711 | 75,982 (0.58%) | 62 (0.08%) | 1 (0.00%) | 17,278 (22.74%) | + |
| 3 | Buea | B02 | Limnogonus cereiventris | 27,361,770 | 161,821 (0.59%) | 53 (0.03%) | 1 (0.00%) | 30,423 (18.80%) | + |
| 4 | Buea | B04 | Limnogonus cereiventris | 12,617,275 | 57,802 (0.46%) | 18 (0.03%) | 0 (0.00%) | 13,120 (22.70%) | + |
| 5 | Buea | B02 | Microvelia sp. 1 | 15,199,889 | 156,452 (1.03%) | 35 (0.02%) | 0 (0.00%) | 6,017 (3.85%) | + |
| 6 | Kumba | K02 | Metrobates sp. 7 | 11,913,069 | 61,477 (0.52%) | 73 (0.12%) | 1 (0.00%) | 29 (0.05%) | - |
| 7 | Kumba | K02 | Metrobates sp. 6 | 11,847,970 | 62,737 (0.53%) | 57 (0.09%) | 0 (0.00%) | 70 (0.11%) | - |
| 8 | Kumba | K02 | Metrobates sp. 8 | 12,711,110 | 66,881 (0.53%) | 68 (0.10%) | 0 (0.00%) | 32 (0.05%) | - |
| 9 | Kumba | K01 | Rhagovelia reitteri | 12,615,049 | 168,912 (1.34%) | 148 (0.09%) | 0 (0.00%) | 106,556 (63.08%) | + |
| 10 | Limbe | L02 | Limnogonus hypoleucus | 13,728,598 | 69,491 (0.51%) | 172 (0.25%) | 89 (0.13%) | 7470 (10.75%) | + |
| 11 | Mbonge | Mb03 | Limnogonus poissoni | 13,897,927 | 87,297 (0.63%) | 67 (0.08%) | 0 (0.00%) | 24,629 (28.21%) | + |
| 12 | Mbonge | Mb03 | Limnogonus poissoni | 13,479,480 | 64,224 (0.48%) | 75 (0.12%) | 0 (0.00%) | 13,345 (20.78%) | + |
| 13 | Mbonge | Mb02 | Limnogonus intermedius | 15,587,895 | 113,606 (0.73%) | 61 (0.05%) | 0 (0.00%) | 9359 (8.24%) | + |
| 14 | Mbonge | Mb04 | Limnogonus poissoni | 14,300,937 | 82,433 (0.58%) | 57 (0.07%) | 0 (0.00%) | 13,412 (16.27%) | + |
| 15 | Mbonge | Mb06 | Rhagovelia sp. 2 | 14,197,736 | 129,978 (0.92%) | 60 (0.05%) | 0 (0.00%) | 66,515 (51.17%) | + |
| 16 | Muyuka | Mu02 | Metrocoris sp. 1 | 14,136,653 | 72,422 (0.51%) | 3598 (4.97%)* | 0 (0.00%) | 142 (0.20%) | - |
| 17 | Muyuka | Mu05 | Eurymetropsis sp. | 14,455,526 | 62,105 (0.43%) | 90 (0.14%) | 0 (0.00%) | 48 (0.08%) | - |
| 18 | Muyuka | Mu01 | Trepobates sp. 5 | 13,054,268 | 57,197 (0.44%) | 37 (0.06%) | 0 (0.00%) | 5326 (9.31%) | - |
| 19 | Muyuka | Mu05 | Rhagovelia sp. 1 | 15,150,283 | 138,234 (0.91%) | 59 (0.04%) | 0 (0.00%) | 5186 (3.75%) | - |
| 20 | Muyuka | Mu05 | Rhagovelia sp. 1 | 14,978,846 | 131,153 (0.88%) | 100 (0.08%) | 0 (0.00%) | 65,354 (49.83%) | + |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Esemu, S.N.; Dong, X.; Kfusi, A.J.; Hartley, C.S.; Ndip, R.N.; Ndip, L.M.; Darby, A.C.; Post, R.J.; Makepeace, B.L. Aquatic Hemiptera in Southwest Cameroon: Biodiversity of Potential Reservoirs of Mycobacterium ulcerans and Multiple Wolbachia Sequence Types Revealed by Metagenomics. Diversity 2019, 11, 225. https://doi.org/10.3390/d11120225
Esemu SN, Dong X, Kfusi AJ, Hartley CS, Ndip RN, Ndip LM, Darby AC, Post RJ, Makepeace BL. Aquatic Hemiptera in Southwest Cameroon: Biodiversity of Potential Reservoirs of Mycobacterium ulcerans and Multiple Wolbachia Sequence Types Revealed by Metagenomics. Diversity. 2019; 11(12):225. https://doi.org/10.3390/d11120225
Chicago/Turabian StyleEsemu, Seraphine N., Xiaofeng Dong, Achah J. Kfusi, Catherine S. Hartley, Roland N. Ndip, Lucy M. Ndip, Alistair C. Darby, Rory J. Post, and Benjamin L. Makepeace. 2019. "Aquatic Hemiptera in Southwest Cameroon: Biodiversity of Potential Reservoirs of Mycobacterium ulcerans and Multiple Wolbachia Sequence Types Revealed by Metagenomics" Diversity 11, no. 12: 225. https://doi.org/10.3390/d11120225
APA StyleEsemu, S. N., Dong, X., Kfusi, A. J., Hartley, C. S., Ndip, R. N., Ndip, L. M., Darby, A. C., Post, R. J., & Makepeace, B. L. (2019). Aquatic Hemiptera in Southwest Cameroon: Biodiversity of Potential Reservoirs of Mycobacterium ulcerans and Multiple Wolbachia Sequence Types Revealed by Metagenomics. Diversity, 11(12), 225. https://doi.org/10.3390/d11120225