Unveiling the History of a Peculiar Weevil-Plant Interaction in South America: A Phylogeographic Approach to Hydnorobius hydnorae (Belidae) Associated with Prosopanche americana (Aristolochiaceae)

Abstract
1. Introduction
2. Materials and Methods
2.1. Study Area and Sample Collection
2.2. DNA Isolation, PCR Amplification and Sequencing
2.3. Haplotype Network, Population Genetic Structure and Genetic Diversity
2.4. Phylogenetic Relationships among Haplotypes
2.5. Demographic History Analysis
2.6. Bayesian Spatio-Temporal Diffusion Analyses
2.7. Paleodistribution Models of the Host Plant Prosopanche americana
3. Results
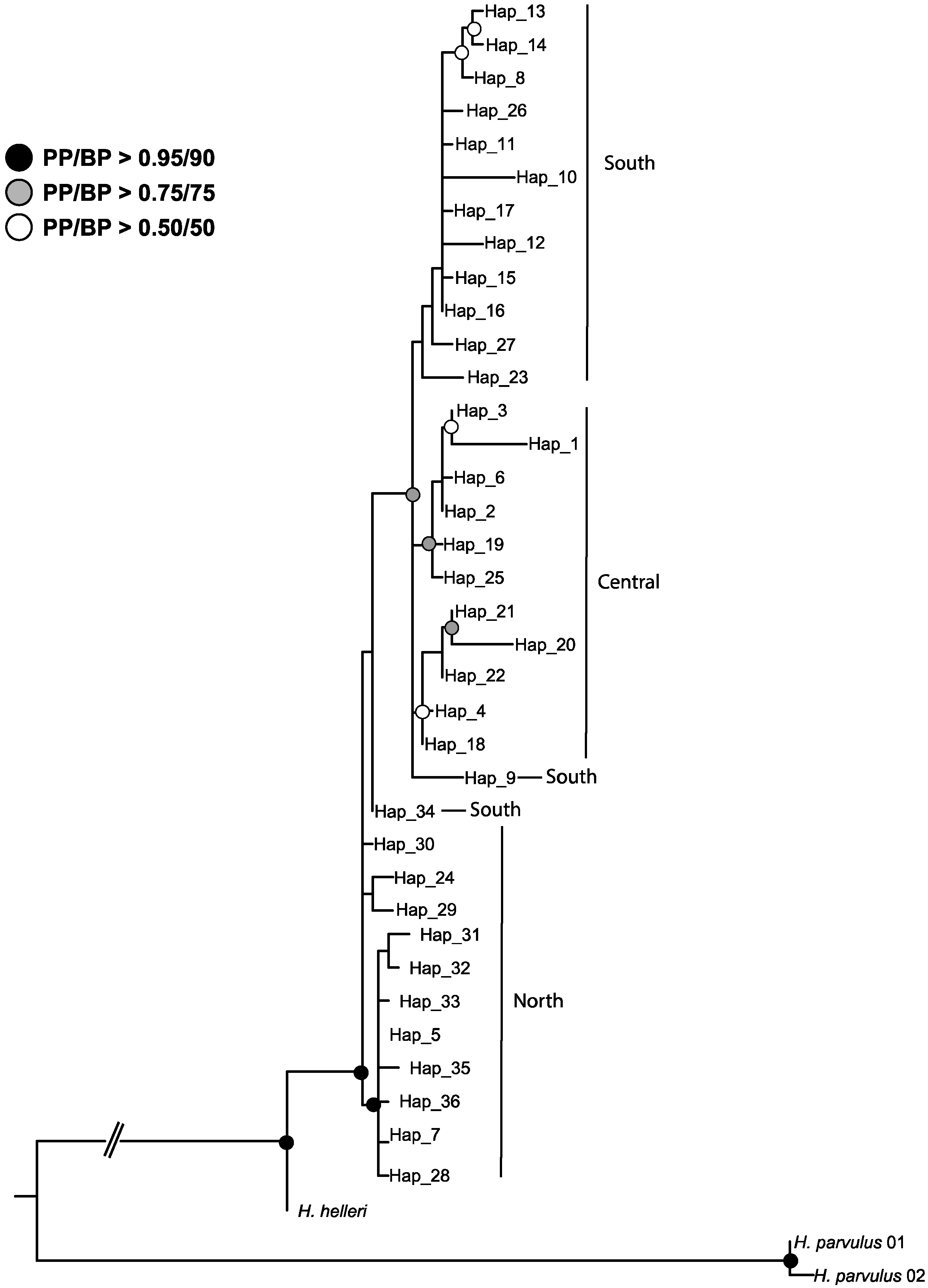
3.1. Strong but Unevenly Distributed Population Structure Across the Range for Hydnorobius hydnorae
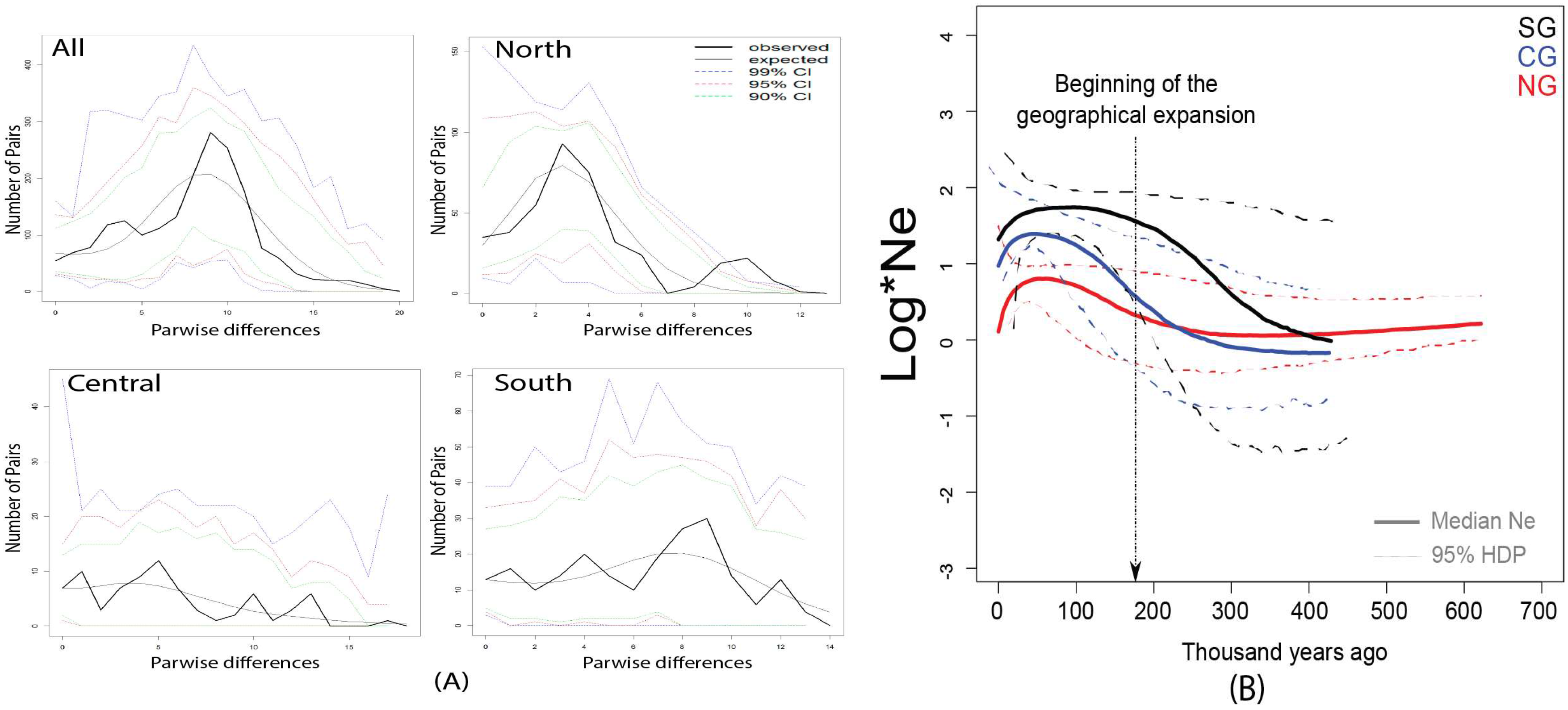
3.2. Weak Signals of Population Expansion for the Hydnorobius Hydnorae Population as a Whole
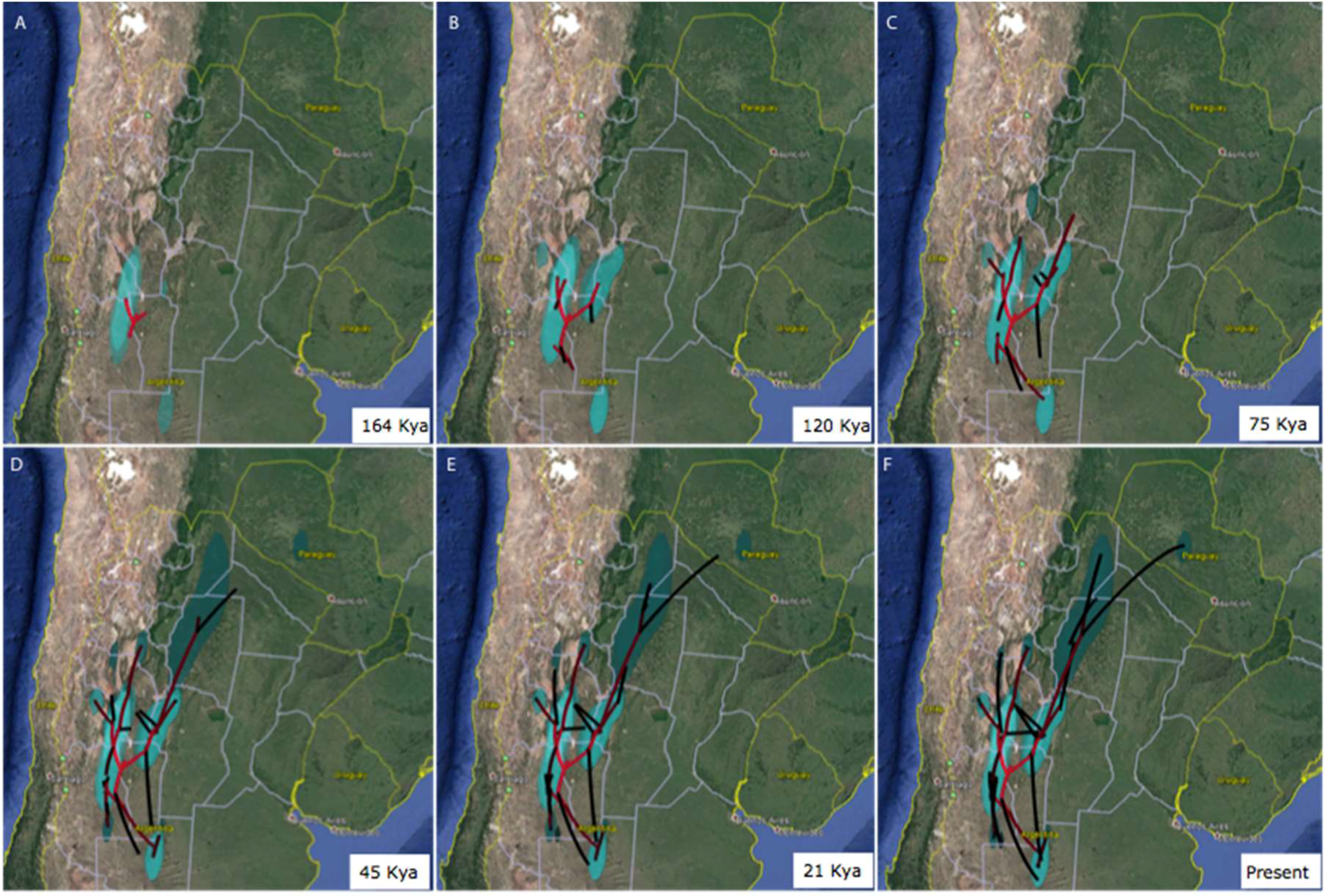
3.3. Area of Origin and North-South Axis of Spatio-Temporal Diffusion for H. hydnorae
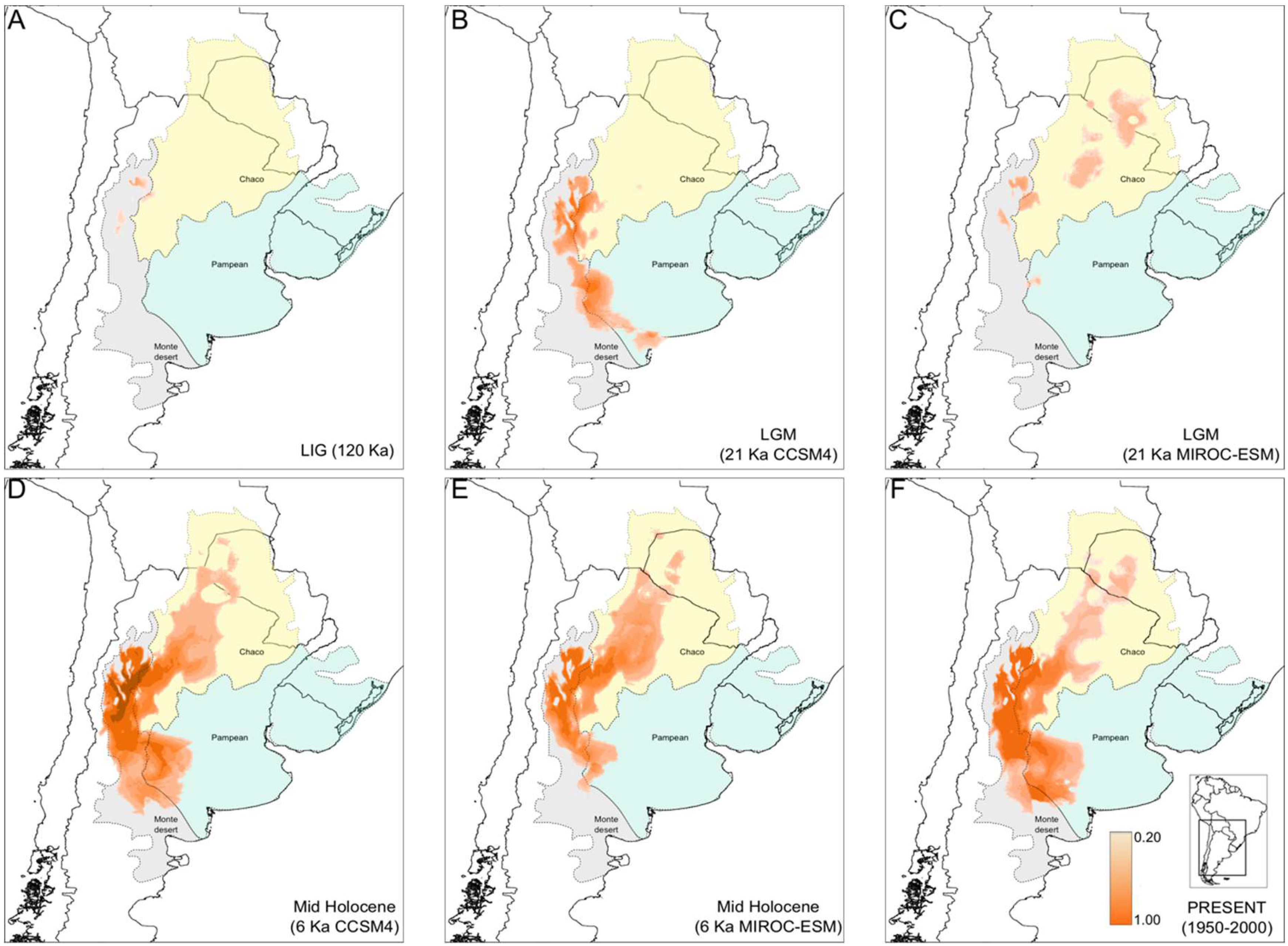
3.4. North-South Range Expansion in Prosopanche Americana, the Host of Hydnorobius hydnorae during 120 Kya
4. Discussion
4.1. Genetic Structure and Geographic Expansions without Major Demographic Change Across the Range of Hydnorobius hydnorae
4.2. Ancestral Weevil Haplotypes and Ancestral Areas for Hydnorobius hydnorae and Its Host Plant
4.3. Concordant Weevil and Host Plant Diffusion-Expansion Patterns
5. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Futuyma, D.J.; Agrawal, A.A. Macroevolution and the biological diversity of plants and herbivores. Proc. Natl. Acad. Sci. USA 2009, 106, 18054–18061. [Google Scholar] [CrossRef] [PubMed]
- Toju, H.; Sota, T. Phylogeography and the geographic cline in the armament of a seed-predatory weevil: Effects of historical events vs. natural selection from the host plant. Mol. Ecol. 2006, 15, 4161–4173. [Google Scholar] [CrossRef] [PubMed]
- De-la-Mora, M.; Piñero, D.; Oyama, K.; Farrell, B.; Magallón, S.; Núñez-Farfán, J. Evolution of Trichobaris (Curculionidae) in relation to host plants: Geometric morphometrics, phylogeny and phylogeography. Mol. Phylogenet. Evol. 2018, 124, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, N.; Kjellberg, F.; Mckey, D.; Hossaert-McKey, M. Phylogeography and historical biogeography of obligate specific mutualisms. In The Biogeography of Host-Parasite Interactions; Oxford University Press: Oxford, UK, 2010; pp. 31–39. ISBN 978-0-19-956134-6. [Google Scholar]
- Gavin, D.G.; Fitzpatrick, M.C.; Gugger, P.F.; Heath, K.D.; Rodríguez-Sánchez, F.; Dobrowski, S.Z.; Hampe, A.; Hu, F.S.; Ashcroft, M.B.; Bartlein, P.J. Climate refugia: Joint inference from fossil records, species distribution models and phylogeography. New Phytol. 2014, 204, 37–54. [Google Scholar] [CrossRef] [PubMed]
- Sosa-Pivatto, M.; Cosacov, A.; Baranzelli, M.C.; Iglesias, M.R.; Espíndola, A.; Sérsic, A.N. Do 120,000 years of plant–pollinator interactions predict floral phenotype divergence in Calceolaria polyrhiza? A reconstruction using species distribution models. Arthropod-Plant Interact. 2017, 11, 351–361. [Google Scholar] [CrossRef]
- Thompson, A.R.; Thacker, C.E.; Shaw, E.Y. Phylogeography of marine mutualists: Parallel patterns of genetic structure between obligate goby and shrimp partners. Mol. Ecol. 2005, 14, 3557–3572. [Google Scholar] [CrossRef] [PubMed]
- Valiente-Banuet, A.; Rumebe, A.V.; Verdú, M.; Callaway, R.M. Modern Quaternary plant lineages promote diversity through facilitation of ancient Tertiary lineages. Proc. Natl. Acad. Sci. USA 2006, 103, 16812–16817. [Google Scholar] [CrossRef] [PubMed]
- Kuschel, G. Oxycorynus missionis spec. nov. from NE Argentina, with key to the South American species of Oxycoryninae (Coleoptera: Belidae). Acta Zool. Lilloana 1995, 43, 45–48. [Google Scholar]
- Kuschel, G. Nemonychidae, Belidae y Oxycorynidae de la fauna chilena, con algunas consideraciones biogeográficas. Investig. Zool. Chile 1959, 5, 229–271. [Google Scholar]
- Ferrer, M.S.; Marvaldi, A.E. New host plant and distribution records for weevils of the genus Hydnorobius (Coleoptera: Belidae). Rev. Soc. Entomol. Argent. 2010, 69, 271–274. [Google Scholar]
- Nickrent, D.L.; Blarer, A.; Qiu, Y.-L.; Soltis, D.E.; Soltis, P.S.; Zanis, M. Molecular data place Hydnoraceae with Aristolochiaceae. Am. J. Bot. 2002, 89, 1809–1817. [Google Scholar] [CrossRef] [PubMed]
- Naumann, J.; Salomo, K.; Der, J.P.; Wafula, E.K.; Bolin, J.F.; Maass, E.; Frenzke, L.; Samain, M.-S.; Neinhuis, C.; Wanke, S. Single-copy nuclear genes place haustorial Hydnoraceae within Piperales and reveal a Cretaceous origin of multiple parasitic angiosperm lineages. PLoS ONE 2013, 8, e79204. [Google Scholar] [CrossRef] [PubMed]
- Massoni, J.; Forest, F.; Sauquet, H. Increased sampling of both genes and taxa improves resolution of phylogenetic relationships within Magnoliidae, a large and early-diverging clade of angiosperms. Mol. Phylogenet. Evol. 2014, 70, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Byng, J.W.; Chase, M.W.; Christenhusz, M.J.; Fay, M.F.; Judd, W.S.; Mabberley, D.J.; Sennikov, A.N.; Soltis, D.E.; Soltis, P.S.; Stevens, P.F. An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG IV. Bot. J. Linn. Soc. 2016, 181, 1–20. [Google Scholar]
- Cocucci, A.E. Estudios en el género Prosopanche (Hydnoraceae). I. Revisión taxonómica. Kurtziana 1965, 2, 53–74. [Google Scholar]
- Cocucci, A.E.; Cocucci, A.A. Prosopanche (Hydnoraceae): Somatic and reproductive structures, biology, systematics, phylogeny and potentialities as a parasitic weed. In Congresos y Jornadas-Junta de Andalucía (España); JA, DGIA: New York, NY, USA, 1996. [Google Scholar]
- Roig-Juñent, S.; Flores, G.; Claver, S.; Debandi, G.; Marvaldi, A. Monte Desert (Argentina): Insect biodiversity and natural areas. J. Arid Environ. 2001, 47, 77–94. [Google Scholar] [CrossRef]
- Roig, F.A.; Roig-Juñent, S.; Corbalán, V. Biogeography of the Monte desert. J. Arid Environ. 2009, 73, 164–172. [Google Scholar] [CrossRef]
- Vogt, C. Composición de la flora vascular del Chaco Boreal, Paraguay III. Dicotyledoneae: Gesneriaceae–Zygophyllaceae. Steviana 2013, 5, 5–40. [Google Scholar]
- Bruch, C. Coleópteros fertilizadores de “Prosopanche Burmeisteri” De Bary. Physis 1923, 7, 82–88. [Google Scholar]
- Marvaldi, A.E. Larval morphology and biology of oxycorynine weevils and the higher phylogeny of Belidae (Coleoptera, Curculionoidea). Zool. Scr. 2005, 34, 37–48. [Google Scholar] [CrossRef]
- Avise, J.C. Phylogeography: The History and Formation of Species; Harvard University Press: Cambridge, MA, USA, 2000; ISBN 0-674-66638-0. [Google Scholar]
- Sérsic, A.N.; Cosacov, A.; Cocucci, A.A.; Johnson, L.A.; Pozner, R.; Avila, L.J.; Sites, J.W.; Morando, M. Emerging phylogeographical patterns of plants and terrestrial vertebrates from Patagonia. Biol. J. Linn. Soc. 2011, 103, 475–494. [Google Scholar] [CrossRef]
- Turchetto-Zolet, A.C.; Pinheiro, F.; Salgueiro, F.; Palma-Silva, C. Phylogeographical patterns shed light on evolutionary process in South America. Mol. Ecol. 2013, 22, 1193–1213. [Google Scholar] [CrossRef] [PubMed]
- Baranzelli, M.C.; Cosacov, A.; Ferreiro, G.; Johnson, L.A.; Sérsic, A.N. Travelling to the south: Phylogeographic spatial diffusion model in Monttea aphylla (Plantaginaceae), an endemic plant of the Monte Desert. PLoS ONE 2017, 12, e0178827. [Google Scholar] [CrossRef] [PubMed]
- Morrone, J.J. Biogeographical regionalisation of the Neotropical region. Zootaxa 2014, 3782, 1–110. [Google Scholar] [CrossRef] [PubMed]
- Rokas, A.; Ladoukakis, E.; Zouros, E. Animal mitochondrial DNA recombination revisited. Trends Ecol. Evol. 2003, 18, 411–417. [Google Scholar] [CrossRef]
- Kraytsberg, Y.; Schwartz, M.; Brown, T.A.; Ebralidse, K.; Kunz, W.S.; Clayton, D.A.; Vissing, J.; Khrapko, K. Recombination of human mitochondrial DNA. Science 2004, 304, 981. [Google Scholar] [CrossRef] [PubMed]
- Avise, J.C.; Arnold, J.; Ball, R.M.; Bermingham, E.; Lamb, T.; Neigel, J.E.; Reeb, C.A.; Saunders, N.C. Intraspecific phylogeography: The mitochondrial DNA bridge between population genetics and systematics. Annu. Rev. Ecol. Syst. 1987, 18, 489–522. [Google Scholar] [CrossRef]
- Crozier, R.H.; Crozier, Y.C. The cytochrome b and ATPase genes of honeybee mitochondrial DNA. Mol. Biol. Evol. 1992, 9, 474–482. [Google Scholar] [PubMed]
- Simon, C.; Frati, F.; Beckenbach, A.; Crespi, B.; Liu, H.; Flook, P. Evolution, weighting, and phylogenetic utility of mitochondrial gene sequences and a compilation of conserved polymerase chain reaction primers. Ann. Entomol. Soc. Am. 1994, 87, 651–701. [Google Scholar] [CrossRef]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. In Nucleic Acids Symposium Series; Information Retrieval Ltd.: London, UK, 1999; Volume 41, pp. 95–98. [Google Scholar]
- Rozas, J.; Sánchez-DelBarrio, J.C.; Messeguer, X.; Rozas, R. DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics 2003, 19, 2496–2497. [Google Scholar] [CrossRef] [PubMed]
- Bandelt, H.-J.; Forster, P.; Röhl, A. Median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 1999, 16, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Leigh, J.W.; Bryant, D. Popart: Full-feature software for haplotype network construction. Methods Ecol. Evol. 2015, 6, 1110–1116. [Google Scholar] [CrossRef]
- Dupanloup, I.; Schneider, S.; Excoffier, L. A simulated annealing approach to define the genetic structure of populations. Mol. Ecol. 2002, 11, 2571–2581. [Google Scholar] [CrossRef] [PubMed]
- Excoffier, L.; Lischer, H.E.L. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar] [CrossRef] [PubMed]
- Flores, G.E.; Roig-Juñent, S. Cladistic and biogeographic analyses of the Neotropical genus Epipedonota Solier (Coleoptera: Tenebrionidae), with conservation considerations. J. N. Y. Entomol. Soc. 2001, 109, 309–336. [Google Scholar] [CrossRef]
- Drummond, A.J.; Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 2007, 7, 214. [Google Scholar] [CrossRef] [PubMed]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef] [PubMed]
- Tracer. Available online: http://tree.bio.ed.ac.uk/software/tracer/ (accessed on 23 March 2018).
- FigTree. Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 23 March 2018).
- Guindon, S.; Dufayard, J.-F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Nei, M.; Kumar, S. Molecular Evolution and Phylogenetics; Oxford University Press: Oxford, UK, 2000; ISBN 0-19-535051-0. [Google Scholar]
- Tajima, F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 1989, 123, 585–595. [Google Scholar] [PubMed]
- Harpending, H.C. Signature of ancient population growth in a low-resolution mitochondrial DNA mismatch distribution. Hum. Biol. 1994, 66, 591–600. [Google Scholar] [PubMed]
- Mckenna, D.D.; Wild, A.L.; Kanda, K.; Bellamy, C.L.; Beutel, R.G.; Caterino, M.S.; Farnum, C.W.; Hawks, D.C.; Ivie, M.A.; Jameson, M.L.; et al. The beetle tree of life reveals that Coleoptera survived end-Permian mass extinction to diversify during the Cretaceous terrestrial revolution. Syst. Entomol. 2015, 40, 835–880. [Google Scholar] [CrossRef]
- Zhang, S.-Q.; Che, L.-H.; Li, Y.; Pang, H.; Ślipiński, A.; Zhang, P. Evolutionary history of Coleoptera revealed by extensive sampling of genes and species. Nat. Commun. 2018, 9, 205. [Google Scholar] [CrossRef] [PubMed]
- Lemey, P.; Rambaut, A.; Welch, J.J.; Suchard, M.A. Phylogeography Takes a Relaxed Random Walk in Continuous Space and Time. Mol. Biol. Evol. 2010, 27, 1877–1885. [Google Scholar] [CrossRef] [PubMed]
- Pybus, O.G.; Tatem, A.J.; Lemey, P. Virus evolution and transmission in an ever more connected world. Proc. R. Soc. B 2015, 282, 20142878. [Google Scholar] [CrossRef] [PubMed]
- Drummond, A.J.; Ho, S.Y.W.; Phillips, M.J.; Rambaut, A. Relaxed Phylogenetics and Dating with Confidence. PLoS Biol. 2006, 4, e88. [Google Scholar] [CrossRef] [PubMed]
- Camargo, A.; Werneck, F.P.; Morando, M.; Sites, J.W.; Avila, L.J. Quaternary range and demographic expansion of Liolaemus darwinii (Squamata: Liolaemidae) in the Monte Desert of Central Argentina using Bayesian phylogeography and ecological niche modelling. Mol. Ecol. 2013, 22, 4038–4054. [Google Scholar] [CrossRef] [PubMed]
- Bielejec, F.; Rambaut, A.; Suchard, M.A.; Lemey, P. SPREAD: Spatial phylogenetic reconstruction of evolutionary dynamics. Bioinformatics 2011, 27, 2910–2912. [Google Scholar] [CrossRef] [PubMed]
- Guisan, A.; Thuiller, W. Predicting species distribution: Offering more than simple habitat models. Ecol. Lett. 2005, 8, 993–1009. [Google Scholar] [CrossRef]
- Graham, C.H.; Ron, S.R.; Santos, J.C.; Schneider, C.J.; Moritz, C. Integrating phylogenetics and environmental niche models to explore speciation mechanisms in dendrobatid frogs. Evolution 2004, 58, 1781–1793. [Google Scholar] [CrossRef] [PubMed]
- Naumann, J.; Der, J.P.; Wafula, E.K.; Jones, S.S.; Wagner, S.T.; Honaas, L.A.; Ralph, P.E.; Bolin, J.F.; Maass, E.; Neinhuis, C. Detecting and characterizing the highly divergent plastid genome of the nonphotosynthetic parasitic plant Hydnora visseri (Hydnoraceae). Genome Biol. Evol. 2016, 8, 345–363. [Google Scholar] [CrossRef] [PubMed]
- WorldClim—Global Climate Data|Free climate data for ecological modeling and GIS. Available online: http://www.worldclim.org/ (accessed on 22 March 2018).
- Hijmans, R.J.; Cameron, S.E.; Parra, J.L.; Jones, P.G.; Jarvis, A. Very high resolution interpolated climate surfaces for global land areas. Int. J. Climatol. 2005, 25, 1965–1978. [Google Scholar] [CrossRef]
- Phillips, S.J.; Anderson, R.P.; Schapire, R.E. Maximum entropy modeling of species geographic distributions. Ecol. Model. 2006, 190, 231–259. [Google Scholar] [CrossRef]
- Anderson, R.P.; Gonzalez, I., Jr. Species-specific tuning increases robustness to sampling bias in models of species distributions: An implementation with Maxent. Ecol. Model. 2011, 222, 2796–2811. [Google Scholar] [CrossRef]
- Otto-Bliesner, B.L.; Marshall, S.J.; Overpeck, J.T.; Miller, G.H.; Hu, A. Simulating Arctic climate warmth and Icefield retreat in the last interglaciation. Science 2006, 311, 1751–1753. [Google Scholar] [CrossRef] [PubMed]
- Schneider, S.; Excoffier, L. Estimation of past demographic parameters from the distribution of pairwise differences when the mutation rates vary among sites: Application to human mitochondrial DNA. Genetics 1999, 152, 1079–1089. [Google Scholar] [PubMed]
- Marvaldi, A.E.; Oberprieler, R.G.; Lyal, C.H.C.; Bradbury, T.; Anderson, R.S. Phylogeny of the Oxycoryninae sensu lato (Coleoptera: Belidae) and evolution of host-plant associations. Invertebr. Syst. 2006, 20, 447–476. [Google Scholar] [CrossRef]
- Werneck, F.P. The diversification of eastern South American open vegetation biomes: Historical biogeography and perspectives. Quat. Sci. Rev. 2011, 30, 1630–1648. [Google Scholar] [CrossRef]
- Ferrer, M.S. Molecular Systematics and Evolution of Belidae, with Special Reference to Oxycoryninae (Coleoptera: Curculionoidea). Ph.D. Thesis, Universidad Nacional de Cuyo, Mendoza, Argentina, 2011. [Google Scholar]
- Ferrer, M.S.; Sequeira, A.S.; Marvaldi, A.E. Host associations in ancient weevils: A phylogenetic perspective on Belidae and Nemonychidae. Diversity, Under preparation.
- Yoke, M.M.; Morando, M.; Avila, L.J.; Sites, J.W., Jr. Phylogeography and genetic structure in the Cnemidophorus longicauda complex (Squamata, Teiidae). Herpetologica 2006, 62, 420–434. [Google Scholar] [CrossRef]
- Morrone, J.J. Biogeographic Areas and Transition Zones of Latin America and the Caribbean Islands Based on Panbiogeographic and Cladistic Analyses of the Entomofauna. Annu. Rev. Entomol. 2006, 51, 467–494. [Google Scholar] [CrossRef] [PubMed]
- Morando, M.; Avila, L.J.; Baker, J.; Sites, J.W., Jr. Phylogeny and phylogeography of the Liolaemus darwinii complex (Squamata: Liolaemidae): Evidence for introgression and incomplete lineage sorting. Evolution 2004, 58, 842–861. [Google Scholar] [CrossRef] [PubMed]
- Masello, J.F.; Quillfeldt, P.; Munimanda, G.K.; Klauke, N.; Segelbacher, G.; Schaefer, H.M.; Failla, M.; Cortés, M.; Moodley, Y. The high Andes, gene flow and a stable hybrid zone shape the genetic structure of a wide-ranging South American parrot. Front. Zool. 2011, 8, 16. [Google Scholar] [CrossRef] [PubMed]
- Vuilleumier, B.S. Pleistocene changes in the fauna and flora of South America. Science 1971, 173, 771–780. [Google Scholar] [CrossRef] [PubMed]
- Markgraf, V. Late and postglacial vegetational and paleoclimatic changes in subantarctic, temperate, and arid environments in Argentina. Palynology 1983, 7, 43–70. [Google Scholar] [CrossRef]
- Rivera, P.C.; González-Ittig, R.E.; Robainas Barcia, A.; Trimarchi, L.I.; Levis, S.; Calderón, G.E.; Gardenal, C.N. Molecular phylogenetics and environmental niche modeling reveal a cryptic species in the Oligoryzomys flavescens complex (Rodentia, Cricetidae). J. Mammal. 2018, 99, 363–376. [Google Scholar] [CrossRef]
- Sánchez, J. Variabilidad Genética, Distribución y Estado de Conservación de Las Poblaciones de Tortugas Terrestres Chelonoidis chilensis (Testudines: Testudinidae) Que Habitan en la República Argentina. Ph.D. Thesis, Universidad Nacional de La Plata, Buenos Aires, Argentina, 2013. [Google Scholar]
- Nullo, F.E.; Stephens, G.C.; Otamendi, J.; Baldauf, P.E. El volcanismo del Terciario superior del sur de Mendoza. Rev. Asoc. Geol. Argent. 2002, 57, 119–132. [Google Scholar]
- Sruoga, P.; Guerstein, P.; Bermudez, A. Riesgo volcánico. In XII Congreso Geológico Argentino; Ramos, V., Ed.; Asociación Geológica Argentina: Mendoza, Argentina, 1993; pp. 659–667. [Google Scholar]
- Ortiz-Jaureguizar, E.; Cladera, G.A. Paleoenvironmental evolution of southern South America during the Cenozoic. J. Arid Environ. 2006, 66, 498–532. [Google Scholar] [CrossRef]
- Neigel, J.E.; Ball, R.M.; Avise, J.C. Estimation of single generation migration distances from geographic variation in animal mitochondrial DNA. Evolution 1991, 45, 423–432. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Anzótegui, L.M.; Garralla, S.S.; Herbst, R. Fabaceae de la Formación El Morterito (Mioceno superior) del valle del Cajón, provincia de Catamarca, Argentina. Ameghiniana 2007, 44, 183–196. [Google Scholar]
- Anzótegui, L.M.; Horn, Y.; Herbst, R. Paleoflora (Fabaceae y Anacardiaceae) de la Formación Andalhuala (Plioceno Inferior), provincia de Catamarca, Argentina. Ameghiniana 2007, 44, 525–535. [Google Scholar]
- Catalano, S.A.; Vilardi, J.C.; Tosto, D.; Saidman, B.O. Molecular phylogeny and diversification history of Prosopis (Fabaceae: Mimosoideae). Biol. J. Linn. Soc. 2008, 93, 621–640. [Google Scholar] [CrossRef]
- Pascual, R.; Ortiz Jaureguizar, E.; Prado, J.L. Land mammals: Paradigm for Cenozoic South American geobiotic evolution. Münch. Geowiss. Abh. 1996, 30, 265–319. [Google Scholar]
- Alberdi, M.T.; Bonnadona, F.P.; Ortiz Jaureguizar, E. Chronological correlation, paleoecology, and paleobiogeography of the late Cenozoic South American Rionegran land-mammal fauna: A review. Rev. Esp. Palent. 1997, 12, 249–255. [Google Scholar]
- Kuschel, G.; Poinar, G.O. Libanorhinus succinus gen. & sp. n. (Coleoptera: Nemonychidae) from Lebanese amber. Insect Syst. Evol. 1993, 24, 143–146. [Google Scholar]
- Kuschel, G.; May, B.M. Discovery of Palophaginae (Coleoptera: Megalopodidae) on Araucaria araucana in Chile and Argentina. N. Z. Entomol. 1996, 19, 1–13. [Google Scholar] [CrossRef]
- Farrell, B.D. “Inordinate fondness” explained: Why are there so many beetles? Science 1998, 281, 555–559. [Google Scholar] [CrossRef] [PubMed]
- Sequeira, A.S.; Normark, B.B.; Farrell, B.D. Evolutionary assembly of the conifer fauna: Distinguishing ancient from recent associations in bark beetles. Proc. R. Soc. Lond. B Biol. Sci. 2000, 267, 2359–2366. [Google Scholar] [CrossRef] [PubMed]
- Sequeira, A.S.; Farrell, B.D. Evolutionary origins of Gondwanan interactions: How old are Araucaria beetle herbivores? Biol. J. Linn. Soc. 2001, 74, 459–474. [Google Scholar] [CrossRef]
- Wilf, P.; Labandeira, C.C.; Johnson, K.R.; Cúneo, N.R. Richness of plant–insect associations in Eocene Patagonia: A legacy for South American biodiversity. Proc. Natl. Acad. Sci. USA 2005, 102, 8944–8948. [Google Scholar] [CrossRef] [PubMed]
- Peterson, A.T.; Soberón, J.; Pearson, R.G.; Anderson, R.P.; Martínez-Meyer, E.; Nakamura, M.; Araújo, M.B. Ecological Niches and Geographic Distributions (MPB-49); Princeton University Press: Princeton, NJ, USA, 2011; ISBN 1-4008-4067-8. [Google Scholar]
- Pease, C.M.; Lande, R.; Bull, J.J. A model of population growth, dispersal and evolution in a changing environment. Ecology 1989, 70, 1657–1664. [Google Scholar] [CrossRef]
- Keane, R.M.; Crawley, M.J. Exotic plant invasions and the enemy release hypothesis. Trends Ecol. Evol. 2002, 17, 164–170. [Google Scholar] [CrossRef]
- Gaume, L.; Matile-Ferrero, D.; Mckey, D. Colony foundation and acquisition of coccoid trophobionts by Aphomomyrmex afer (Formicinae): Co-dispersal of queens and phoretic mealybugs in an ant-plant-homopteran mutualism? Insectes Soc. 2000, 47, 84–91. [Google Scholar] [CrossRef]
- Waltari, E.; Perkins, S.L. In the hosts footsteps? Ecological niche modeling and its utility in predicting parasite distributions. In The Biolgeography of Host-Parasite Interactions; Oxford University Press: Oxford, UK, 2010; pp. 145–155. ISBN 978-0-19-956134-6. [Google Scholar]
- Hoberg, E.P.; Brooks, D.R. A macroevolutionary mosaic: Episodic host-switching, geographical colonization and diversification in complex host–parasite systems. J. Biogeogr. 2008, 35, 1533–1550. [Google Scholar] [CrossRef]
- Krauss, J.; Steffan-Dewenter, I.; Tscharntke, T. Landscape occupancy and local population size depends on host plant distribution in the butterfly Cupido minimus. Biol. Conserv. 2004, 120, 355–361. [Google Scholar] [CrossRef]
- León-Cortés Jorge, L.; Lennon Jack, J.; Thomas Chris, D. Ecological dynamics of extinct species in empty habitat networks. 2. The role of host plant dynamics. Oikos 2003, 102, 465–477. [Google Scholar] [CrossRef]
- Thomas, C.D.; Bodsworth, E.J.; Wilson, R.J.; Simmons, A.D.; Davies, Z.G.; Musche, M.; Conradt, L. Ecological and evolutionary processes at expanding range margins. Nature 2001, 411, 577–581. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Province and Locality Name | Population Code | Coordinates | N | |
|---|---|---|---|---|
| North | La Rioja: San Ramón | LSR | S 30° 22.002′; W 66° 52.002′ | 4 |
| San Luis: Quines | SLQUI | S 32° 17.472′; W 65° 50.982′ | 5 | |
| Córdoba: Chancaní | CCH | S 31° 22.584′; W 65° 28.812′ | 4 | |
| Córdoba: Árbol blanco | CAB | S 30° 9.06′; W 64° 4.404′ | 5 | |
| Paraguay: Hayes | PHAY | S 23° 4.818′; W 59° 13.272′ | 3 | |
| Santiago del Estero: Aurora-Huayapampa | SAUHU | S 27° 25.416′; W 64° 16.686′ | 5 | |
| Salta: La Unión | SLU | S 23° 45.918′; W 63° 4.914′ | 1 | |
| Chaco: Taco Pozo | CHTP | S 25° 40.74′; W 63° 7.8′ | 2 | |
| Central | Catamarca: Pio Brizuela | CBR | S 27° 49.878′; W 66° 12,16′ | 3 |
| Catamarca: Tinogasta | CTI | S 28° 4.99′; W 67° 34.002′ | 4 | |
| San Juan: Bermejo | SBE | S 31° 31.998′; W 67° 24′ | 3 | |
| San Juan: Huaco | SHU | S 30° 9′; W 68° 34.998′ | 3 | |
| South | Mendoza: Divisadero | MDI | S 33° 11.4′; W 67° 51′ | 5 |
| Mendoza: Reserva de la Biósfera Ñacuñán | MNA | S 34° 3′; W 67° 57′ | 4 | |
| Mendoza: Paso del Loro | MPD | S 35° 39.672′; W 67° 33,492′ | 5 | |
| La Pampa: Chacharramendi | LPCHA | S 37° 24.372′; W 65° 18.39′ | 1 | |
| La Pampa: El Durazno | LPDZO | S 36° 40.448′; W 65° 17.346′ | 3 | |
| La Pampa: La Maruja | LPMAR | S 35° 37.62′; W 64° 50.418′ | 3 |
| Source of Variation | % of Variation | Fixation Indices (Φ-Statistics) |
|---|---|---|
| Among all localities without hierarchical levels | 53.45 | ΦST = 0.5345 * |
| Within Localities | 46.55 | |
| Among regional groups as single large populations | 47.09 | ΦST = 0.4709 * |
| Within regional groups | 52.91 | |
| Among SAMOVA proposed groupings | 44.88 | ΦCT = 0.4488 * |
| Among localities within groups | 15.32 | ΦSC = 0.2780 * |
| Within localities | 39.80 | ΦST = 0.6020 * |
| CAB | 0.398 * | 0 | |||||||||||||||
| PHAY | 0.436 * | 0.793 * | 0 | ||||||||||||||
| SAUHU | −0.013 | 0.553 * | 0.681 * | 0 | |||||||||||||
| SLU | −0.220 | 0.715 | 1 | 0 | 0 | ||||||||||||
| CHTP | 0.178 | 0.779 * | 1 | 0.286 | 0 | 0 | |||||||||||
| SLQUI | 0.06 | 0.581 | 0.68 | 0.2 | 0.335 | 0.508 | 0 | ||||||||||
| LSR | −0.239 | 0.33 | 0.353 * | 0 | −0.2 | 0.152 | 0.06 | 0 | |||||||||
| CBR | 0.13 | 0.033 | 0.032 | 0.045 | 0.098 | 0.05 | 0.04 | 0.152 | 0 | ||||||||
| CTI | 0.07 | 0.013 | 0 | 0.02 | 0 | 0 | 0.018 | 0.076 | 0.047 | 0 | |||||||
| SHU | 0.1 | 0.031 | 0.028 | 0.043 | 0.09 | 0.047 | 0.039 | 0.119 | 0.145 | 0.065 | 0 | ||||||
| SBE | 0.2 | 0.033 | 0.023 | 0.053 | 0.066 | 0.036 | 0.044 | 0.223 | 0.212 | 0.118 | 0.194 | 0 | |||||
| MDI | 0.27 | 0.1 | 0.129 | 0.132 | 0.511 | 0.177 | 0.105 | 0.304 | 0.223 | 0.184 | 0.199 | 0.556 | 0 | ||||
| MPD | 0.17 | 0.06 | 0.068 | 0.078 | 0.15 | 0.087 | 0.065 | 0.186 | 0.144 | 0.124 | 0.14 | 0.386 | −0.154 | 0 | |||
| MNA | 0.12 | 0.05 | 0.055 | 0.056 | 0.113 | 0.065 | 0.05 | 0.146 | 0.106 | 0.056 | 0.101 | 0.132 | 0.136 | 0.215 | 0 | ||
| LPDZO | 0.4 | 0.09 | 0.08 | 0.115 | 0.369 | 0.114 | 0.091 | 0.533 | 0.161 | 0.091 | 0.151 | 0.407 | −0.051 | 0.005 | 0.065 | 0 | |
| LPMAR | 0.09 | 0.028 | 0.017 | 0.034 | 0.033 | 0.02 | 0.03 | 0.121 | 0.08 | 0.022 | 0.072 | 0.109 | 0.036 | 0.054 | 0.116 | −0.059 | 0 |
| LPCHA | 0.18 | 0.022 | 0 | 0.034 | 0 | 0 | 0.031 | 0.213 | 0.197 | 0 | 0.107 | 0.083 | −0.014 | 0.3 | 0.446 | 0.141 | 0.686 |
| Tajima’s Ds | Fu’s Fs | |||||||
|---|---|---|---|---|---|---|---|---|
| Area/Population | n | h | K | π | Ds | p Value | Fs | p Value |
| LSR | 4 | 0.8333 | 2.6667 | 0.0351 | −0.3145 | 0.533 | 0.8114 | 0.568 |
| SLQUI | 5 | 0.8000 | 2.0000 | 0.0048 | −1.1240 | 0.071 | −1.0116 | 0.114 |
| CCH | 4 | 1.0000 | 5.1667 | 0.0123 | −0.5281 | 0.453 | −0.4805 | 0.205 |
| CAB | 5 | 0.4000 | 1.6000 | 0.0038 | −1.0938 | 0.080 | 2.2024 | 0.830 |
| PHAY | 3 | 0.3333 | 0.0000 | 0.0000 | 0.0000 | 1 | N/A | N/A |
| SAUHU | 5 | 0.4000 | 1.8000 | 0.0043 | 1.5727 | 0.965 | 2.4285 | 0.859 |
| SLU | 1 | N/A | N/A | N/A | N/A | N/A | N/A | N/A |
| CHTP | 2 | 0.5000 | 0 | 0 | 0 | 1 | N/A | N/A |
| North | 29 | 0.4482 | 3.8276 | 0.0091 | −1.0018 | 0.161 | −3.7314 | 0.086 |
| CBR | 3 | 1.0000 | 4.0000 | 0.0526 | 0.0000 | 0.551 | 2.3031 | 0.543 |
| CTI | 4 | 0.5000 | 2.5000 | 0.0329 | −0.7968 | 0.166 | 2.5980 | 0.859 |
| SBE | 3 | 0.6667 | 0.6667 | 0.0088 | 0.0000 | 1 | 0.0000 | N. A. |
| SHU | 3 | 0.8333 | 12.0833 | 0.1590 | −1.4104 | 0.078 | 3.0688 | 0.092 |
| Central | 13 | 0.6923 | 5.4359 | 0.0129 | −0.8445 | 0.208 | −1.5152 | 0.201 |
| MDI | 5 | 0.9000 | 3.2000 | 0.0421 | −0.8173 | 0.149 | −1.0124 | 0.106 |
| MNA | 4 | 0.7000 | 1.4000 | 0.0184 | 0.0000 | 0.948 | 1.0609 | 0.561 |
| MPD | 5 | 0.8000 | 1.6000 | 0.0210 | 0.6990 | 0.785 | 0.2764 | 0.523 |
| LPCHA | 1 | N/A | N/A | N/A | N/A | N/A | N/A | N/A |
| LPDZO | 3 | 1 | 6.0000 | 0.0142 | 0.0000 | 1 | 0.5878 | 0.400 |
| LPMAR | 3 | 0.6667 | 2.0000 | 0.0047 | 0.0000 | 1 | 1.6094 | 0.701 |
| South | 22 | 0.6667 | 6.2905 | 0.0149 | −0.9651 | 0.172 | −3.4286 | 0.007 |
| ALL | 63 | 0.5714 | 7.8218 | 0.0185 | −1.5263 | 0.111 | −16.064 | <0.05 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sequeira, A.S.; Rocamundi, N.; Ferrer, M.S.; Baranzelli, M.C.; Marvaldi, A.E. Unveiling the History of a Peculiar Weevil-Plant Interaction in South America: A Phylogeographic Approach to Hydnorobius hydnorae (Belidae) Associated with Prosopanche americana (Aristolochiaceae). Diversity 2018, 10, 33. https://doi.org/10.3390/d10020033
Sequeira AS, Rocamundi N, Ferrer MS, Baranzelli MC, Marvaldi AE. Unveiling the History of a Peculiar Weevil-Plant Interaction in South America: A Phylogeographic Approach to Hydnorobius hydnorae (Belidae) Associated with Prosopanche americana (Aristolochiaceae). Diversity. 2018; 10(2):33. https://doi.org/10.3390/d10020033
Chicago/Turabian StyleSequeira, Andrea S., Nicolás Rocamundi, M. Silvia Ferrer, Matias C. Baranzelli, and Adriana E. Marvaldi. 2018. "Unveiling the History of a Peculiar Weevil-Plant Interaction in South America: A Phylogeographic Approach to Hydnorobius hydnorae (Belidae) Associated with Prosopanche americana (Aristolochiaceae)" Diversity 10, no. 2: 33. https://doi.org/10.3390/d10020033
APA StyleSequeira, A. S., Rocamundi, N., Ferrer, M. S., Baranzelli, M. C., & Marvaldi, A. E. (2018). Unveiling the History of a Peculiar Weevil-Plant Interaction in South America: A Phylogeographic Approach to Hydnorobius hydnorae (Belidae) Associated with Prosopanche americana (Aristolochiaceae). Diversity, 10(2), 33. https://doi.org/10.3390/d10020033