
Synthesis and Characterization of cis-/trans-(±)-3-Alkyl-3,4-dihydro-6,7-dimethoxy-1-oxo-1H-isochromene-4-carboxylic Acids
Abstract
1. Introduction
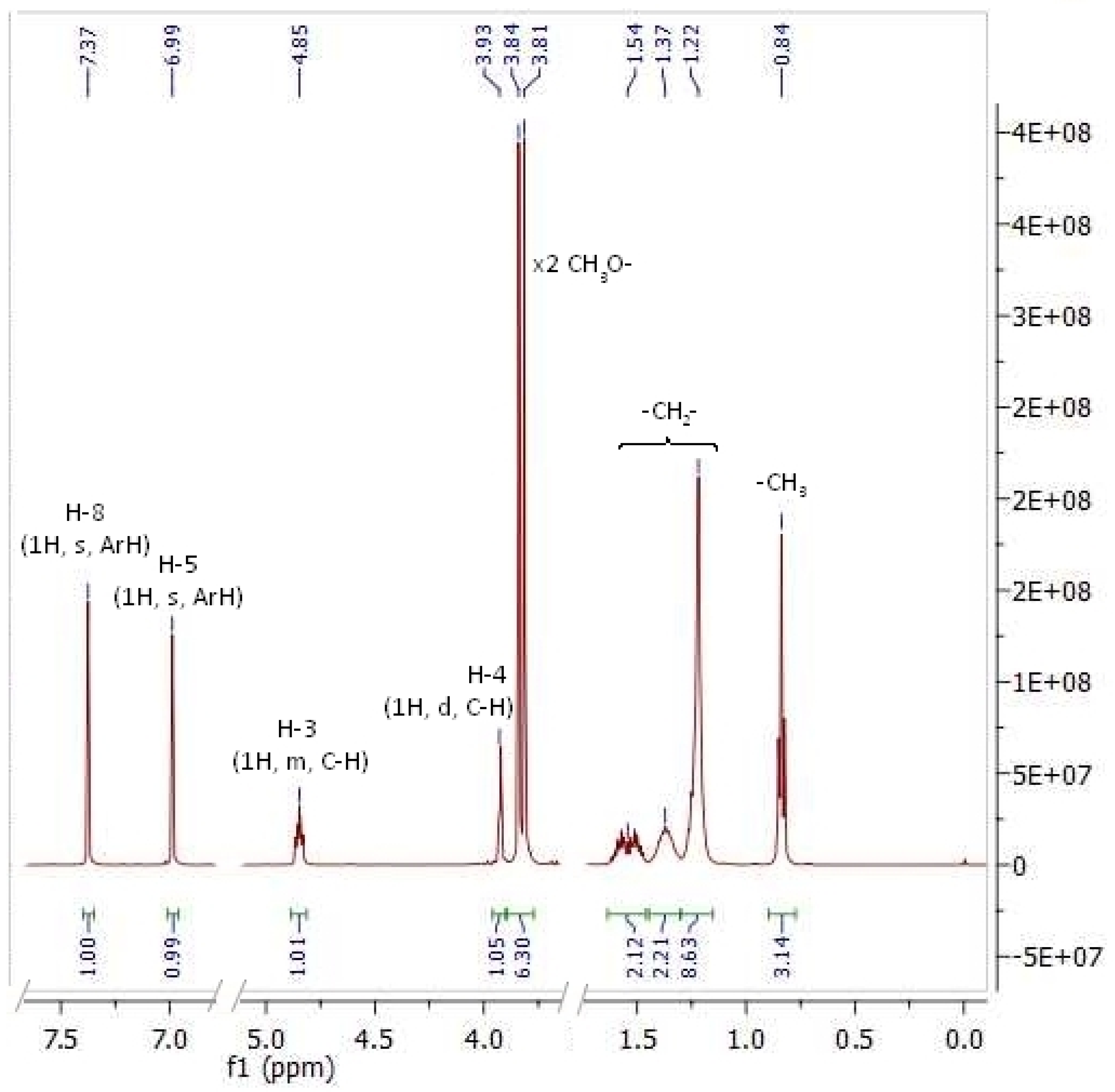
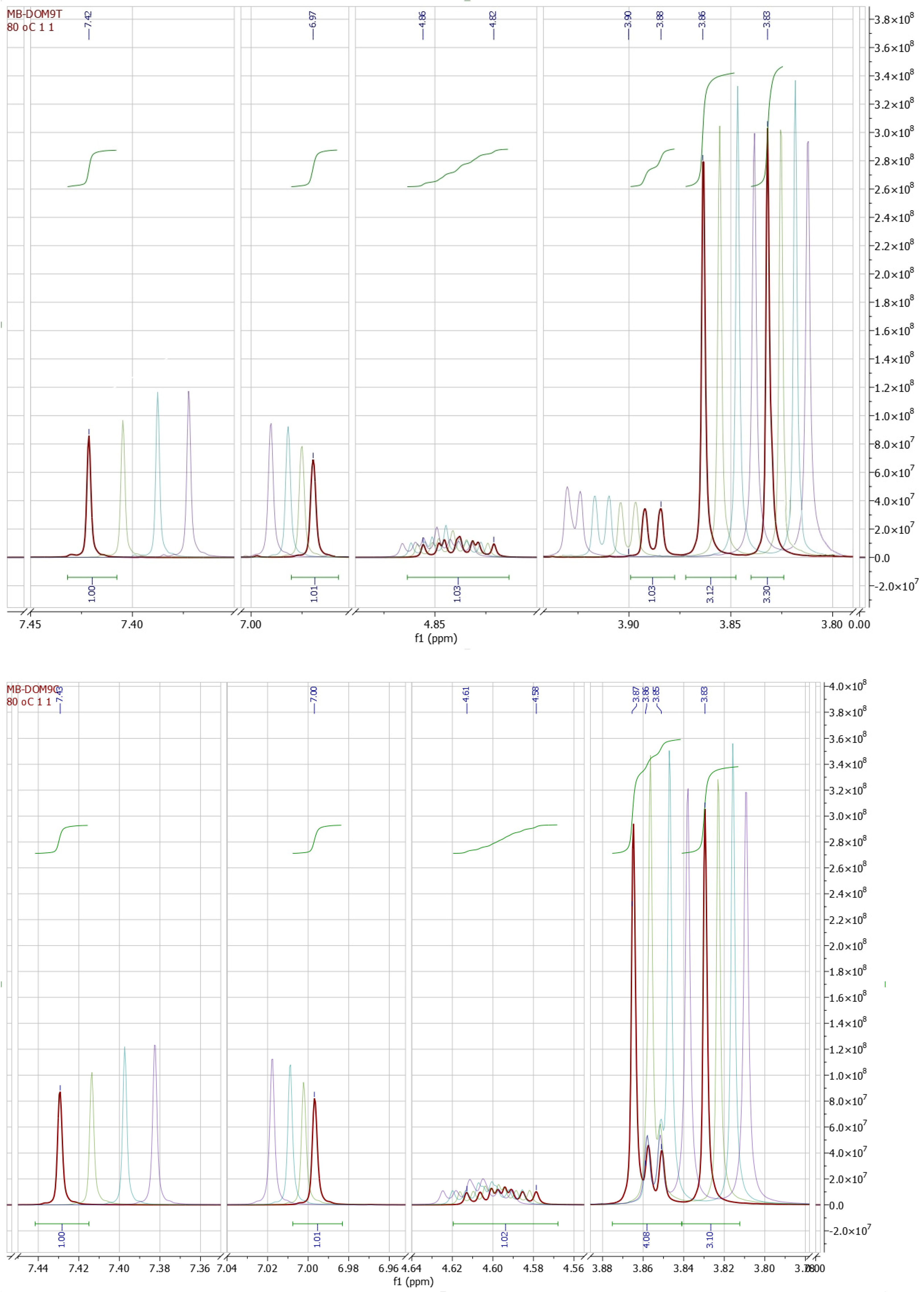
2. Results and Discussion
3. Materials and Methods
3.1. General
3.2. Synthesis
3.2.1. cis- and trans-(±)-3-Heptyl-3,4-dihydro-6,7-dimethoxy-1-oxo-1H-isochromene-4-carboxylic Acids (1)
3.2.2. cis- and trans-(±)-3,4-Dihydro-6,7-dimethoxy-3-nonyl-1-oxo-1H-isochromene-4-carboxylic Acids (2)
3.2.3. cis- and trans-(±)-3-Decyl-3,4-dihydro-6,7-dimethoxy-1-oxo-1H-isochromene-4-carboxylic Acids (3)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Hill, R. Naturally Occurring Isocoumarins. Fortschr. Chem. Org. Naturst. 1986, 49, 1–78. [Google Scholar]
- Napolitano, E. The synthesis of isocoumarins over the last decade. A review. Org. Prep. Proced. Int. 1997, 29, 631–664. [Google Scholar]
- Orfali, R.; Perveen, S.; AlAjmI, M.F.; Ghaffar, S.; Rehman, M.T.; AlanzI, A.R.; Gamea, S.B.; Essa Khwayri, M. Antimicrobial Activity of Dihydroisocoumarin Isolated from Wadi Lajab Sediment-Derived Fungus Penicillium chrysogenum: In Vitro and In Silico Study. Molecules 2022, 27, 3630. [Google Scholar] [CrossRef]
- Hussain, M.; Hussain, M.T.; Rama, N.H.; Hameed, S.; Malik, A.; Khan, K.M. Synthesis and antimicrobial activities of some isocoumarin and dihydroisocoumarin derivatives. Nat. Prod. Res. 2003, 17, 207–214. [Google Scholar] [CrossRef]
- Devienne, K.F.; Raddi, M.S.G.; Coelho, R.G.; Vilegas, W. Structure-antimicrobial activity of some natural isocoumarins and their analogues. Phytomedicine 2005, 12, 378–381. [Google Scholar] [CrossRef]
- Nozawa, K.; Yamada, M.; Tsuda, Y.; Kawai, K.; Nakajima, S. Antifungal activity of oosponol, oospolactone, phyllodulcin, hydrangenol, and some other related compounds. Chem. Pharm. Bull. 1981, 29, 2689–2691. [Google Scholar]
- Kovacs, T.; Sonnenbichler, I.; Sonnenbichler, J. Total synthesis of the toxin oosponol and of structural analogues and investigation of their antibiotic activities. Justus. Liebigs. Ann. Chem. 1997, 4, 773–777. [Google Scholar]
- Matsuda, H.; Shimoda, H.; Yamahara, J.; Yoshikawa, M. Immunomodulatory activity of thunberginol A and related compounds isolated from Hydrangeae Dulcis Folium on splenocyte proliferation activated by mitogens. Biorg. Med. Chem. Lett. 1998, 8, 215–220. [Google Scholar]
- Hamada, Y.; Hara, O.; Kawai, A.; Kohno, Y.; Shioiri, T. Efficient total synthesis of AI-77-B, A gastroprotective substance from bacillus pumilus AI-77. Tetrahedron 1991, 47, 8635–8652. [Google Scholar] [CrossRef]
- Matsuda, H.; Shimoda, H.; Yoshikawa, M. Structure-requirements of isocoumarins, phthalides and stilbenes from Hydrangeae Dulcis Folium for inhibitory activity on histamine release from rat peritoneal mast cells. Bioorg. Med. Chem. Lett. 1999, 7, 1445–1450. [Google Scholar]
- Shimojima, Y.; Shirai, T.; Baba, T.; Hayashi, H. 1H-2-Benzopyran derivatives, microbial products with pharmacological activity. Conversion into orally active derivatives with antiinflammatory and antiulcer activities. J. Med. Chem. 1985, 28, 3–9. [Google Scholar] [PubMed]
- Patel, S.K.; Murat, K.; Py, S.; Vallee, Y. Asymmetric total synthesis and stereochemical elucidation of the antitumor agent PM-94128. Org. Lett. 2003, 5, 4081–4084. [Google Scholar] [PubMed]
- Rauwald, H.W.; Maucher, R.; Dannhardt, G.; Kuchta, K. Dihydroisocoumarins, Naphthalenes, and Further Polyketides from Aloe vera and A. plicatilis: Isolation, Identification and Their 5-LOX/COX-1 Inhibiting Potency. Molecules 2021, 26, 4223. [Google Scholar] [CrossRef]
- Sukandar, E.R.; Kaennakam, S.; Raab, P.; Nöst, X.; Rassamee, K.; Bauer, R.; Siripong, P.; Ersam, T.; Tip-pyang, S.; Chavasiri, W. Cytotoxic and Anti-Inflammatory Activities of Dihydroisocoumarin and Xanthone Derivatives from Garcinia picrorhiza. Molecules 2021, 26, 6626. [Google Scholar] [CrossRef] [PubMed]
- Koopklang, K.; Choodej, S.; Hantanong, S.; Intayot, R.; Jungsuttiwong, S.; Insumran, Y.; Ngamrojanavanich, N.; Pudhom, K. Anti-Inflammatory Properties of Oxygenated Isocoumarins and Xanthone from Thai Mangrove-Associated Endophytic Fungus Setosphaeria rostrata. Molecules 2024, 29, 603. [Google Scholar] [CrossRef]
- Saikia, P.; Gogoi, S. Isocoumarin: General aspects and recent advances in the synthesis. Adv. Synth. Catal. 2018, 360, 2063–2075. [Google Scholar]
- Hussain, H.; Jabeen, F.; Krohn, K.; Al-Harrasi, A.; Ahmad, M.; Mabood, F.; Shah, A.; Badshah, A.; Ur Rehman, N.; Green, I.; et al. Antimicrobial activity of two mellein derivatives isolated from an endophytic fungus. Med. Chem. Res. 2015, 24, 2111–2114. [Google Scholar]
- Choudhary, M.; Musharraf, S.; Mukhmoor, T.; Shaheen, F.; Ali, S.; Ur Rahman, A. Isolation of Bioactive Compounds from Aspergillus terreus. Z. Naturforsch. 2004, 59, 324–328. [Google Scholar]
- Bogdanov, M.; Palamareva, M. cis/trans-Isochromanones. DMAP induced cycloaddition of homophthalic anhydride and aldehydes. Tetrahedron 2004, 60, 2525–2530. [Google Scholar]
- Scriven, E.F.V. 4-Dialkylaminopyridines: Super acylation and alkylation catalysts. Chem. Soc. Rev. 1983, 12, 129–161. [Google Scholar]
- Bogdanov, M.G.; Svinyarov, I.V.; Ivanova, B.B.; Spiteller, M. Synthesis, spectroscopic and structural study of trans- and cis-(±)-3-phenyl-4-(pyrrolidine-1-carbonyl)-isochroman-1-ones. Spectrochim. Acta A 2010, 77, 902–907. [Google Scholar]
- Bogdanov, M.; Todorov, I.; Manolova, P.; Cheshmedzhieva, D.; Palamareva, M. Configuration and conformational equilibrium of (±)-trans-1-oxo-3-thiophen-2-yl-isochroman-4-carboxylic acid methyl ester. Tetrahedron Lett. 2004, 45, 8383–8386. [Google Scholar]
- Miliovsky, M.; Svinyarov, I.; Mitrev, Y.; Evstatieva, Y.; Nikolova, D.; Chochkova, M.; Bogdanov, M. A novel one-pot synthesis and preliminary biological activity evaluation of cis-restricted polyhydroxy stilbenes incorporating protocatechuic acid and cinnamic acid fragments. Eur. J. Med. Chem. 2013, 66, 185–192. [Google Scholar]
- Miliovsky, M.; Svinyarov, I.; Prokopova, E.; Batovska, D.; Stoyanov, S.; Bogdanov, M. Synthesis and Antioxidant Activity of Polyhydroxylated trans-Restricted 2-Arylcinnamic Acids. Molecules 2015, 20, 2555–2575. [Google Scholar] [CrossRef] [PubMed]
- Karplus, M. Contact ElectronSpin Coupling of Nuclear Magnetic Moments. J. Chem. Phys. 1959, 30, 11–15. [Google Scholar]
- Karplus, M. Vicinal Proton Coupling in Nuclear Magnetic Resonance. J. Am. Chem. Soc. 1963, 85, 2870–2871. [Google Scholar]


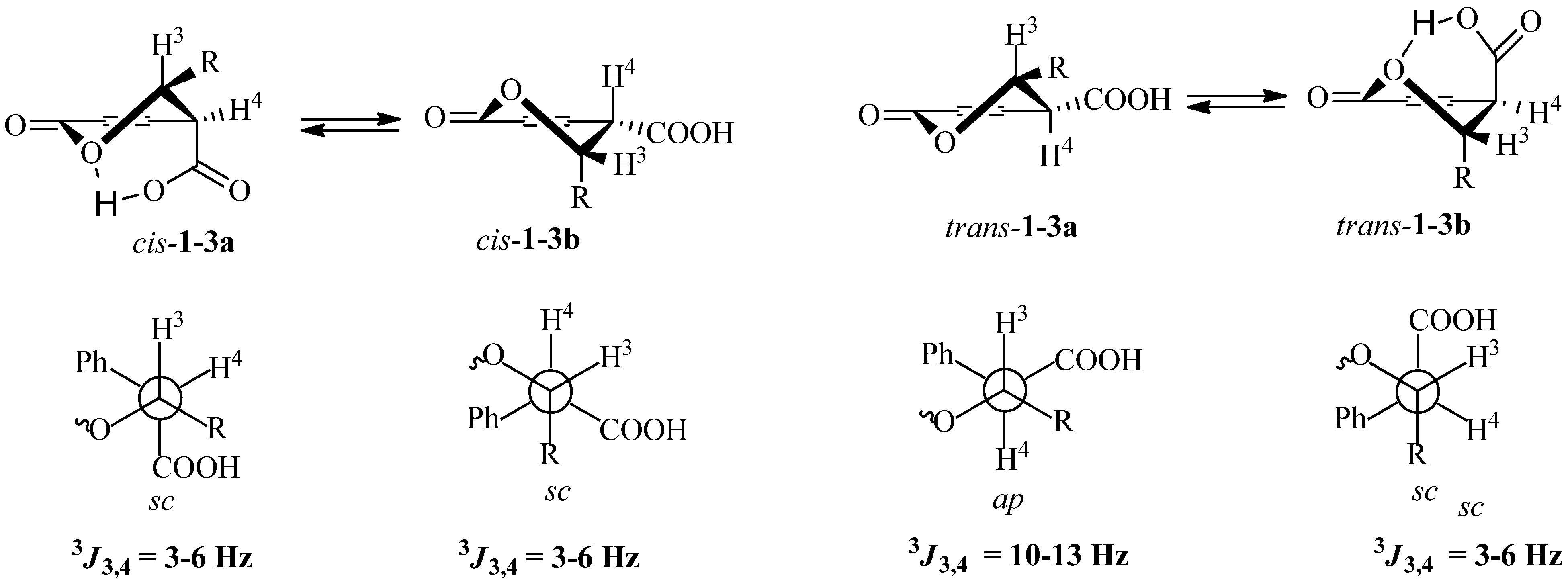

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| R | Yield, % | cis, % | trans, % | |
|---|---|---|---|---|
| 1 | –C7H15 | 92 | 40 | 60 |
| 2 | –C9H19 | 85 | 40 | 60 |
| 3 | –C10H21 | 77 | 41 | 59 |
| Compd. | Chemical Shifts (ppm), Multiplicity, and Coupling Constants (Hz) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| H-8, s | H-5, s | H-4, d | H-3, m * | 3J3,4 | ||||||
| CDCl3 | DMSO | CDCl3 | DMSO | CDCl3 | DMSO | CDCl3 | DMSO | CDCl3 | DMSO | |
| cis-1 | 7.57 | 7.39 | 6.73 | 7.02 | 3.78 | 3.86 | 4.59 (4.62–4.57) | 4.62 (4.65–4.58) | 3.2 | 3.3 |
| cis-2 | 7.58 | 7.39 | 6.73 | 7.02 | 3.73 | 3.85 | 4.59 (4.61–4.56) | 4.61 (4.66–4.56) | 3.0 | 3.3 |
| cis-3 | 7.58 | 7.38 | 6.73 | 7.02 | 3.73 | 3.86 | 4.58 (4.60–4.55) | 4.61 (4.66–4.55) | 3.2 | 3.3 |
| Average (cis) | 7.58 | 7.39 | 6.73 | 7.02 | 3.75 | 3.86 | 4.59 | 4.61 | 3.1 | 3.3 |
| trans-1 | 7.57 | 7.37 | 6.73 | 6.99 | 3.74 | 3.93 | 4.91 (4.95–4.87) | 4.85 (4.88–4.81) | 4.4 | 3.3 |
| trans-2 | 7.57 | 7.38 | 6.73 | 6.99 | 3.78 | 3.92 | 4.91 (4.94–4.88) | 4.85 (4.89–4.80) | 4.5 | 3.3 |
| trans-3 | 7.58 | 7.37 | 6.73 | 6.99 | 3.78 | 3.92 | 4.92 (4.95–4.88) | 4.84 (4.89–4.79) | 4.5 | 3.3 |
| Average (trans) | 7.57 | 7.37 | 6.73 | 6.99 | 3.77 | 3.92 | 4.91 | 4.85 | 4.5 | 3.3 |
| Δδ | 0.01 | 0.02 | n.a. | 0.03 | 0.02 | 0.06 | 0.32 | 0.24 | 1.4 | n.a. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stoyanova, S.; Bogdanov, M.G. Synthesis and Characterization of cis-/trans-(±)-3-Alkyl-3,4-dihydro-6,7-dimethoxy-1-oxo-1H-isochromene-4-carboxylic Acids. Molbank 2025, 2025, M1988. https://doi.org/10.3390/M1988
Stoyanova S, Bogdanov MG. Synthesis and Characterization of cis-/trans-(±)-3-Alkyl-3,4-dihydro-6,7-dimethoxy-1-oxo-1H-isochromene-4-carboxylic Acids. Molbank. 2025; 2025(2):M1988. https://doi.org/10.3390/M1988
Chicago/Turabian StyleStoyanova, Savina, and Milen G. Bogdanov. 2025. "Synthesis and Characterization of cis-/trans-(±)-3-Alkyl-3,4-dihydro-6,7-dimethoxy-1-oxo-1H-isochromene-4-carboxylic Acids" Molbank 2025, no. 2: M1988. https://doi.org/10.3390/M1988
APA StyleStoyanova, S., & Bogdanov, M. G. (2025). Synthesis and Characterization of cis-/trans-(±)-3-Alkyl-3,4-dihydro-6,7-dimethoxy-1-oxo-1H-isochromene-4-carboxylic Acids. Molbank, 2025(2), M1988. https://doi.org/10.3390/M1988