
Synthesis of a 2-(2-Trifluoroethoxyphenyl)oxazoline



{kind=link}
Abstract
1. Introduction
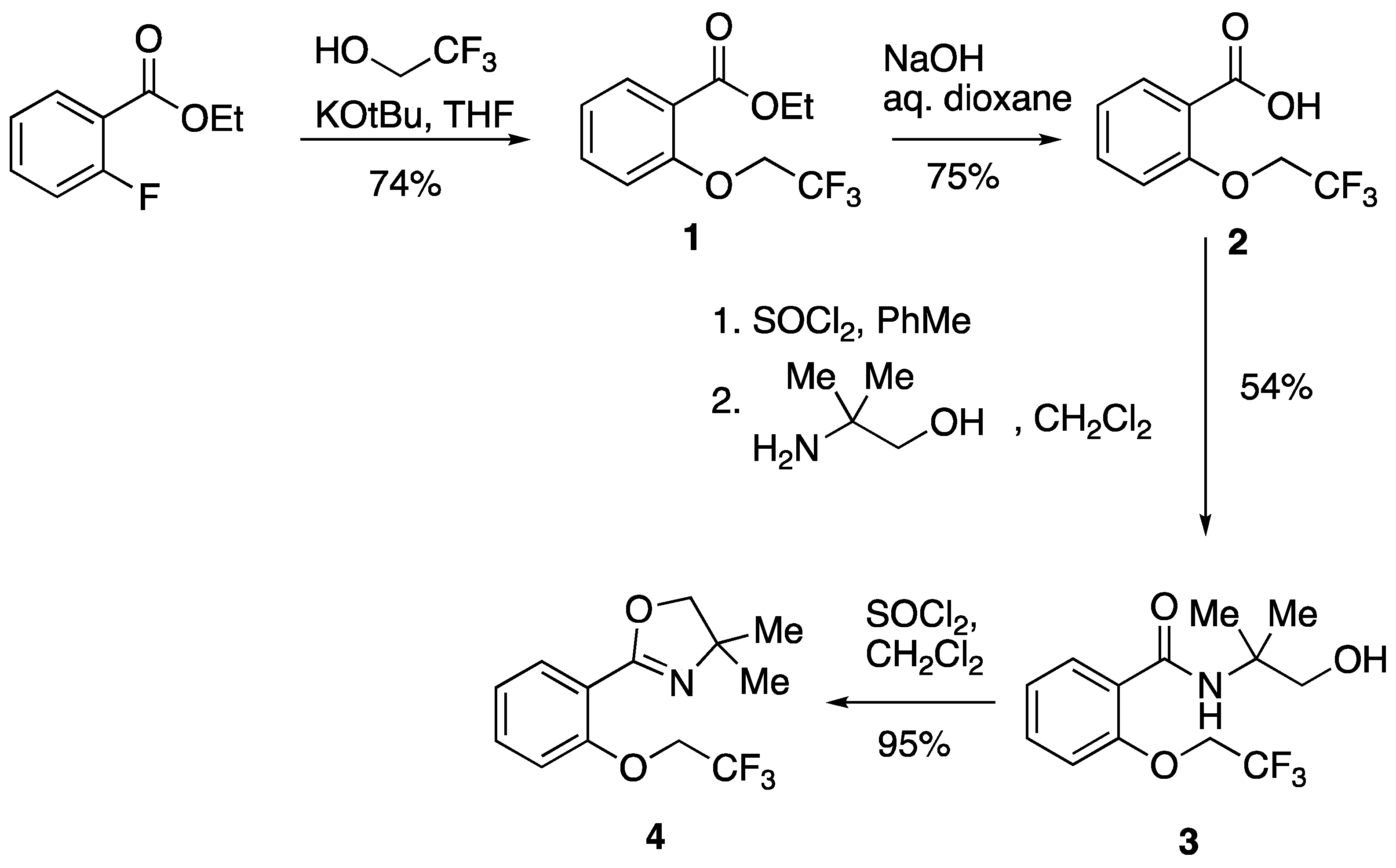
2. Results
3. Experimental Section
3.1. General Experimental Details
3.2. Ethyl 2-(2,2,2-Trifluoroethoxy)benzoate 1
3.3. 2-(2,2,2-Trifluoroethoxy)benzoic acid 2
3.4. N-(1-Hydroxy-2-methylpropan-2-yl)-2-(2,2,2-trifluoroethoxy)benzamide 3
3.5. 4,4-Dimethyl-2-(2-(2,2,2-trifluoroethoxy)phenyl)-4,5-dihydrooxazole 4
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Meyers, A.I.; Mihelich, E.D. The synthetic utility of 2-oxazolines. Angew. Chem. Int. Ed. Engl. 1976, 15, 270–281. [Google Scholar] [CrossRef]
- Meyers, A.I.; Reumann, M. The synthetic utility of oxazolines in aromatic substitution. Tetrahedron 1985, 41, 837–860. [Google Scholar] [CrossRef]
- Gant, T.G.; Meyers, A.I. The chemistry of 2-oxazolines (1985-present). Tetrahedron 1994, 50, 2297–2360. [Google Scholar] [CrossRef]
- Aitken, R.A.; Harper, A.D.; Slawin, A.M.Z. Base-induced cyclisation of ortho-substituted 2-phenyloxazolines to give 3-aminobenzofurans and related heterocycles. Synlett 2017, 28, 1738–1742. [Google Scholar] [CrossRef]
- Aitken, R.A.; Harper, A.D.; Slawin, A.M.Z. Rationalisation of patterns of competing reactivity by X-ray structure determination: Reaction of isomeric (benzyloxythienyl)oxazolines with a base. Molecules 2021, 26, 7690. [Google Scholar] [CrossRef] [PubMed]
- Aitken, R.A.; Harper, A.D.; Inwood, R.A.; Slawin, A.M.Z. Access to diarylmethanols by Wittig rearrangement of ortho-, meta-, and para-benzyloxy-N-butylbenzamides. J. Org. Chem. 2022, 87, 4692–4701. [Google Scholar] [CrossRef] [PubMed]
- Uneyama, K.; Katagiri, T.; Amii, H. α-Trifluoromethylated carbanion synthons. Acc. Chem. Res. 2008, 41, 817–829. [Google Scholar] [CrossRef] [PubMed]
- Aitken, R.A.; Murray, L. New gas-phase cascade reactions of stabilized phosphorus ylides leading to ring-fused indoles and to quinolines. J. Org. Chem. 2008, 73, 9781–9783. [Google Scholar] [CrossRef] [PubMed]
- Idoux, J.P.; Madenwald, M.L.; Garcia, B.S.; Chu, D.-L. Aromatic fluoroalkylation vis direct displacement of a nitro or fluoro group. J. Org. Chem. 1985, 50, 1876–1878. [Google Scholar] [CrossRef]
- Priepke, H.; Doods, H.; Kuelzer, R.; Pfau, R.; Stenkamp, D.; Pelcman, B.; Roenn, R. New compounds. PCT Int. Appl. WO 2012/022793 A1, 23 February 2012. [Google Scholar]
- Mendel, A. Polyfluoroalkoxy-substituted aromatic carboxylic acids and esters and salts thereof. US Patent 3766247, 16 October 1973. [Google Scholar]
- Huang, S.-T.; Hsei, I.-J.; Chen, C. Synthesis and anticancer evaluation of bis(benzimidazoles), bis(benzoxazoles), and benzothiazoles. Bioorg. Med. Chem. 2006, 14, 6106–6119. [Google Scholar] [CrossRef] [PubMed]
- Liverton, N.; Kuduk, S.D.; Luo, Y.; Meng, N.; Yu, T. Piperidine, oxadiazole and thiadiazole orexin receptor antagonists. PCT Int. Appl. WO 2016/069510 A1, 6 May 2016. [Google Scholar]
- Pourkaveh, R.; Svatunek, D.; Schnürch, M. Palladium-catalyzed ortho alkoxylation of oxazoline derivatives: An avenue to reach meta-substituted electron-rich arenes exploiting oxazoline as a removeable directing group. ACS Omega 2024, 9, 44224–44232. [Google Scholar] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aitken, R.A.; Harper, A.D. Synthesis of a 2-(2-Trifluoroethoxyphenyl)oxazoline. Molbank 2025, 2025, M1989. https://doi.org/10.3390/M1989
Aitken RA, Harper AD. Synthesis of a 2-(2-Trifluoroethoxyphenyl)oxazoline. Molbank. 2025; 2025(2):M1989. https://doi.org/10.3390/M1989
Chicago/Turabian StyleAitken, R. Alan, and Andrew D. Harper. 2025. "Synthesis of a 2-(2-Trifluoroethoxyphenyl)oxazoline" Molbank 2025, no. 2: M1989. https://doi.org/10.3390/M1989
APA StyleAitken, R. A., & Harper, A. D. (2025). Synthesis of a 2-(2-Trifluoroethoxyphenyl)oxazoline. Molbank, 2025(2), M1989. https://doi.org/10.3390/M1989