
Synthesis of N-p-Fluorothiosemicarbazone and of Bis(N-p-Fluorophenylthiourea): Crystal Structure and Conformational Analysis of N,N′-Bis(4-Fluorophenyl)Hydrazine-1,2-Bis(Carbothioamide)

 , ,
, ,  , , , and
, , , and
Abstract
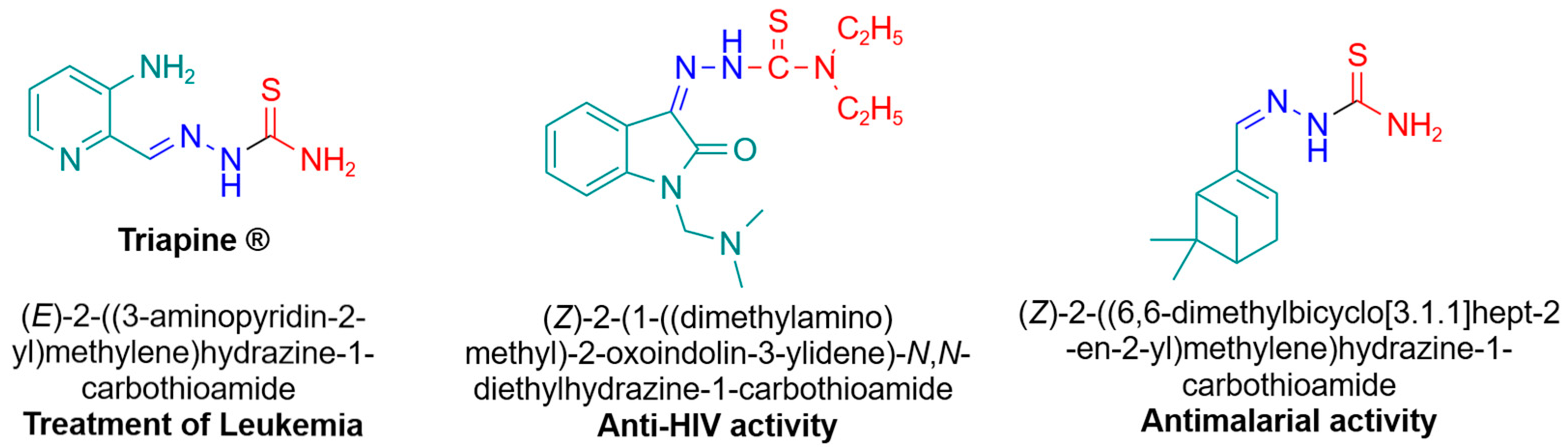
1. Introduction
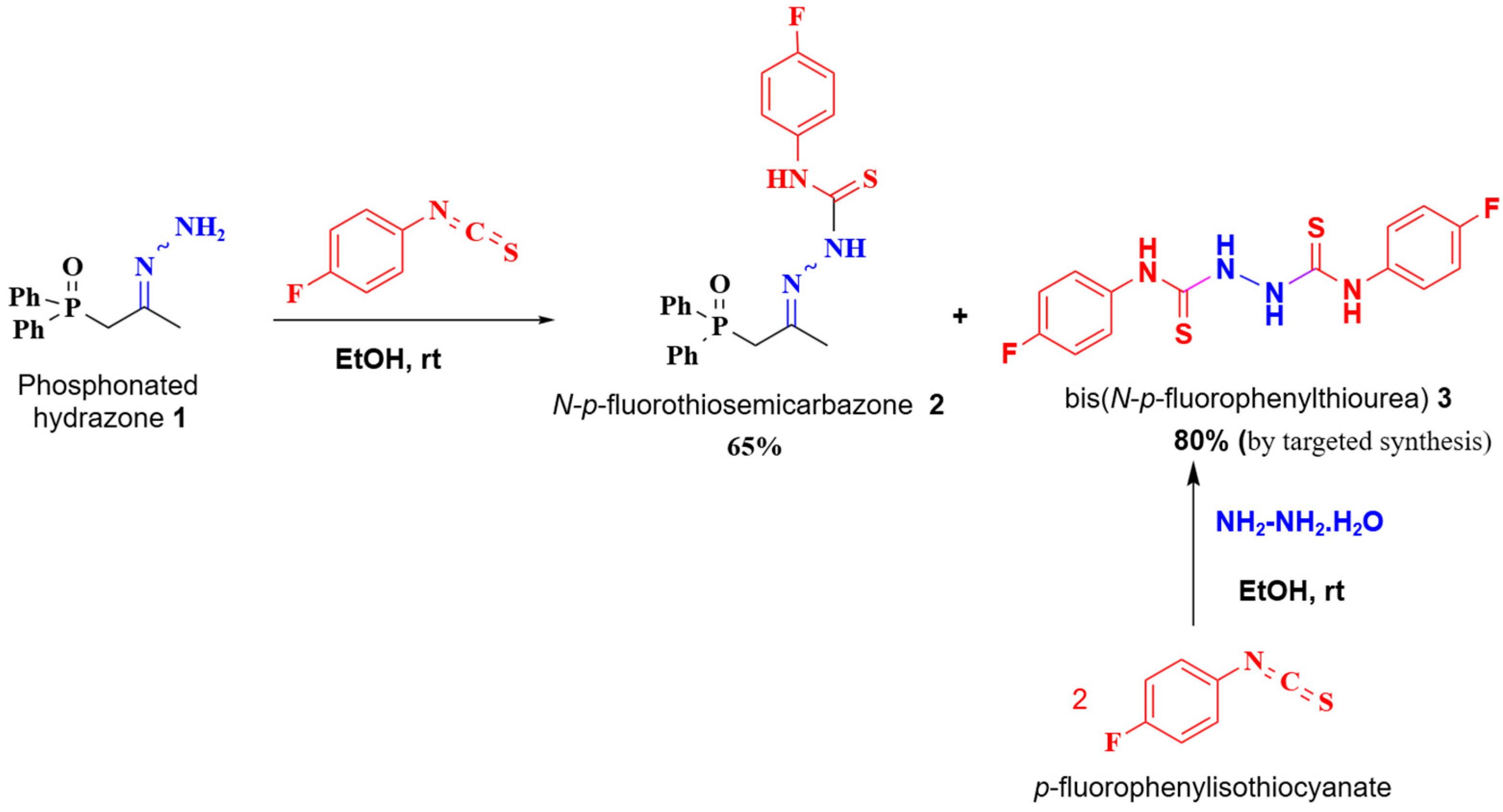
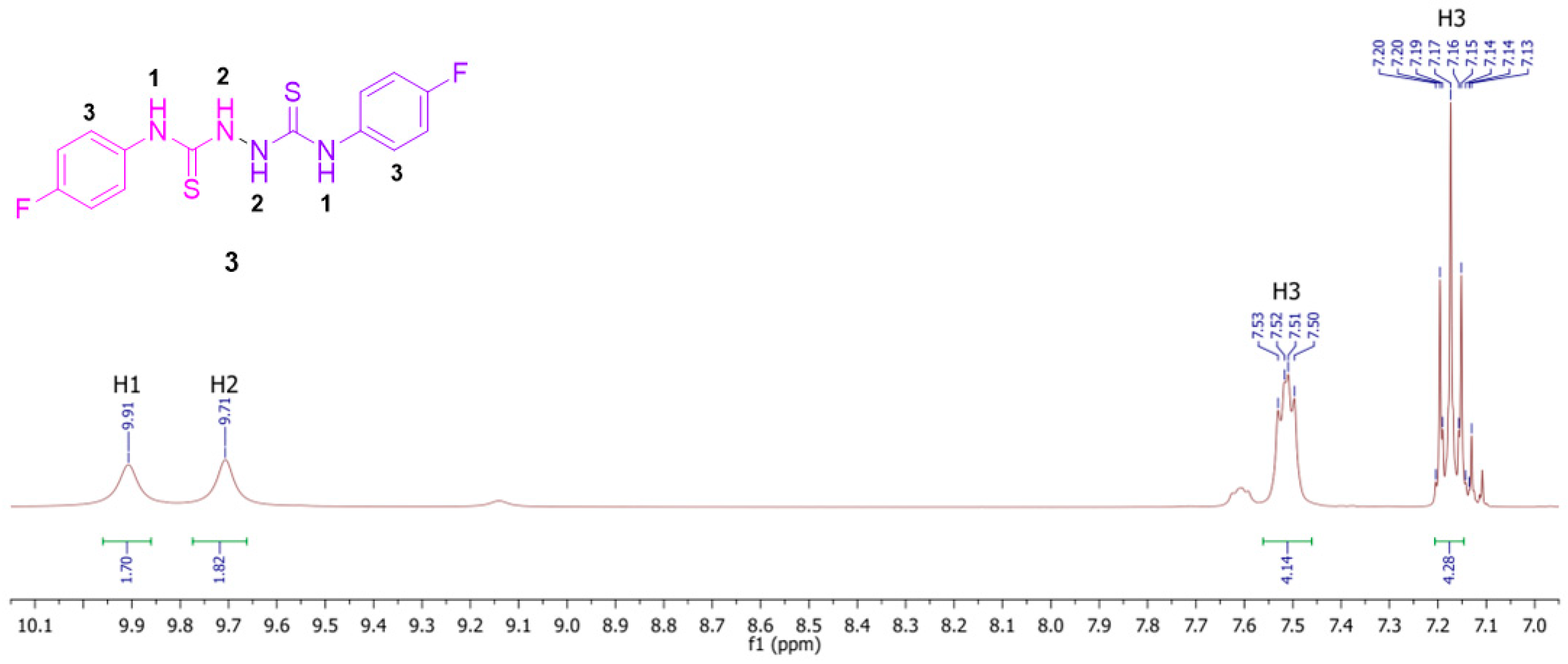
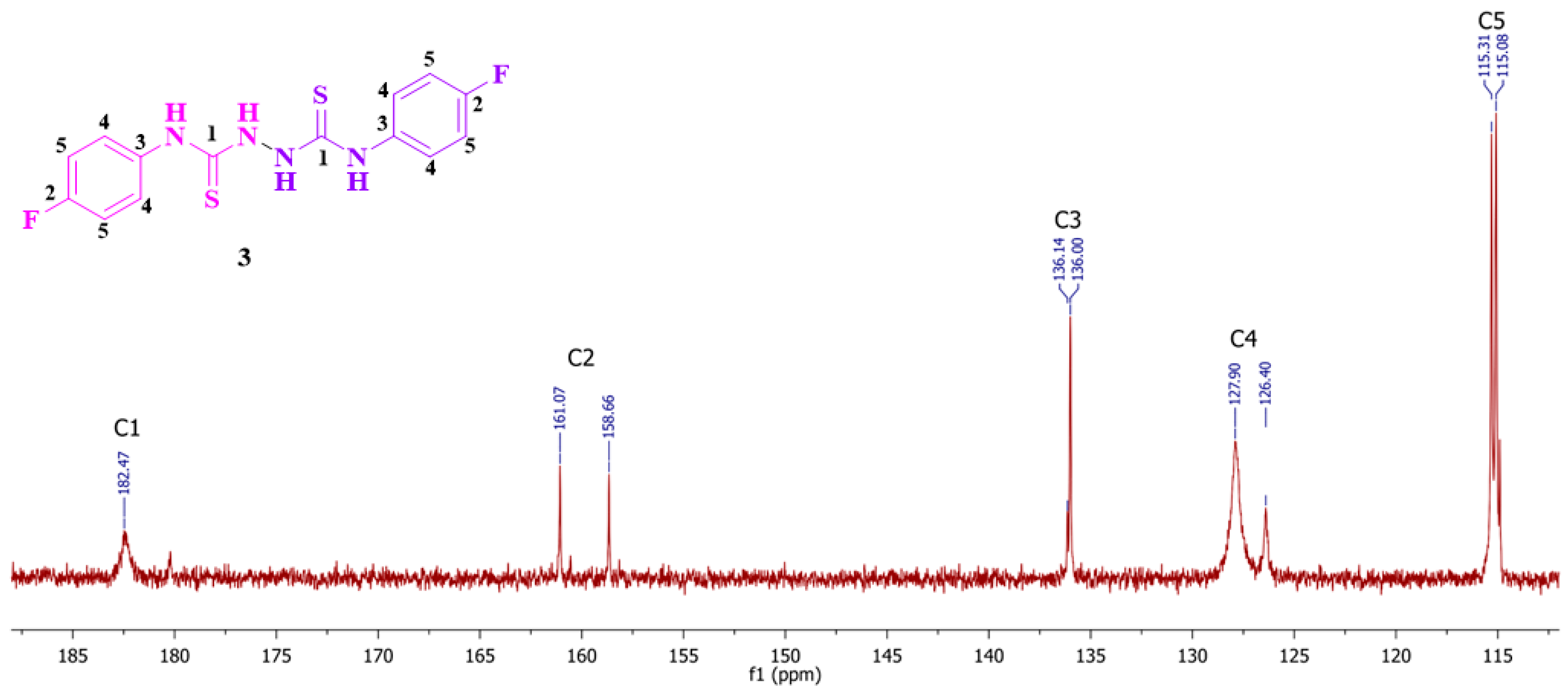
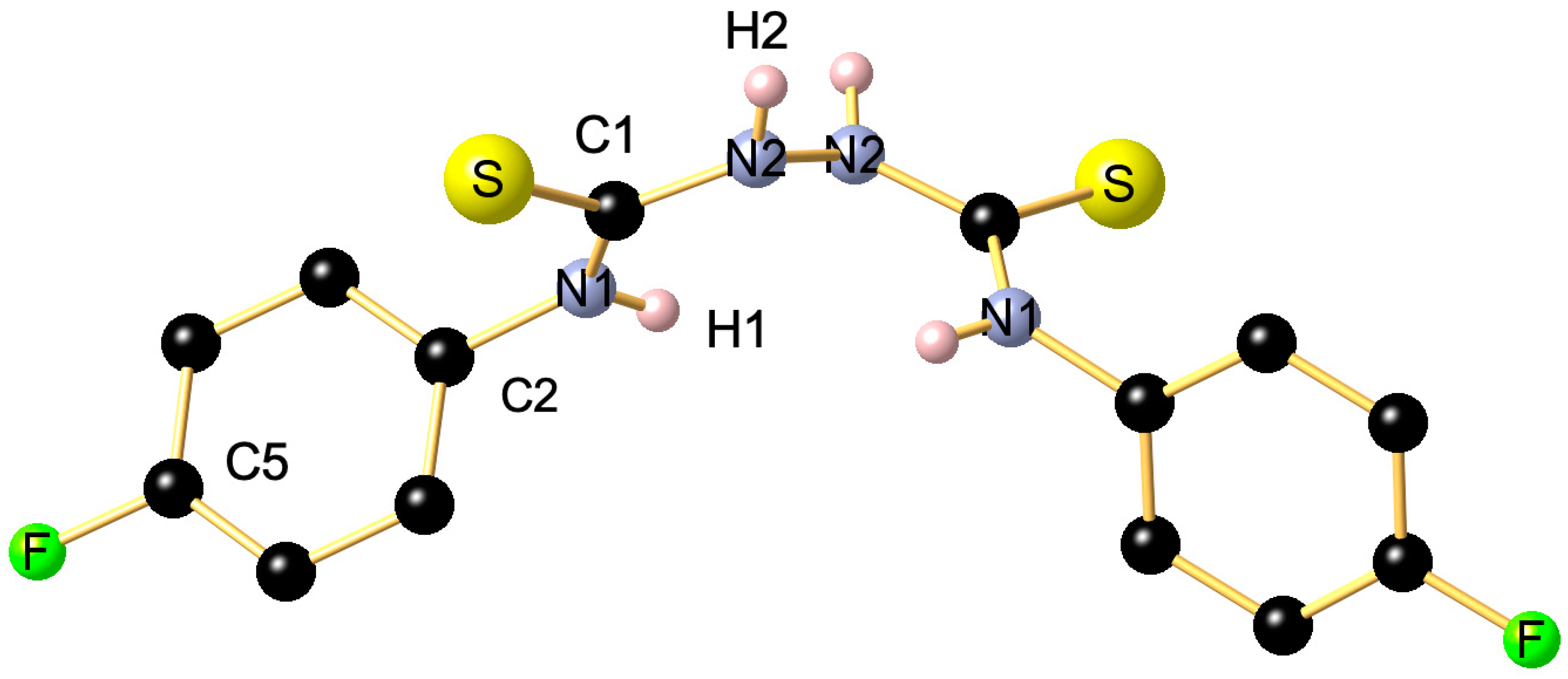
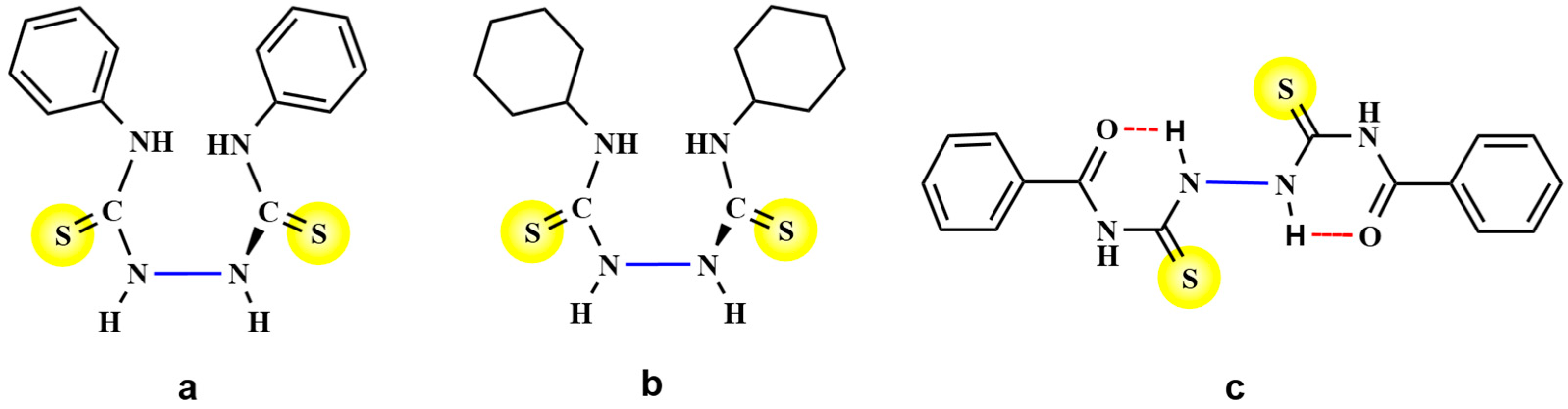
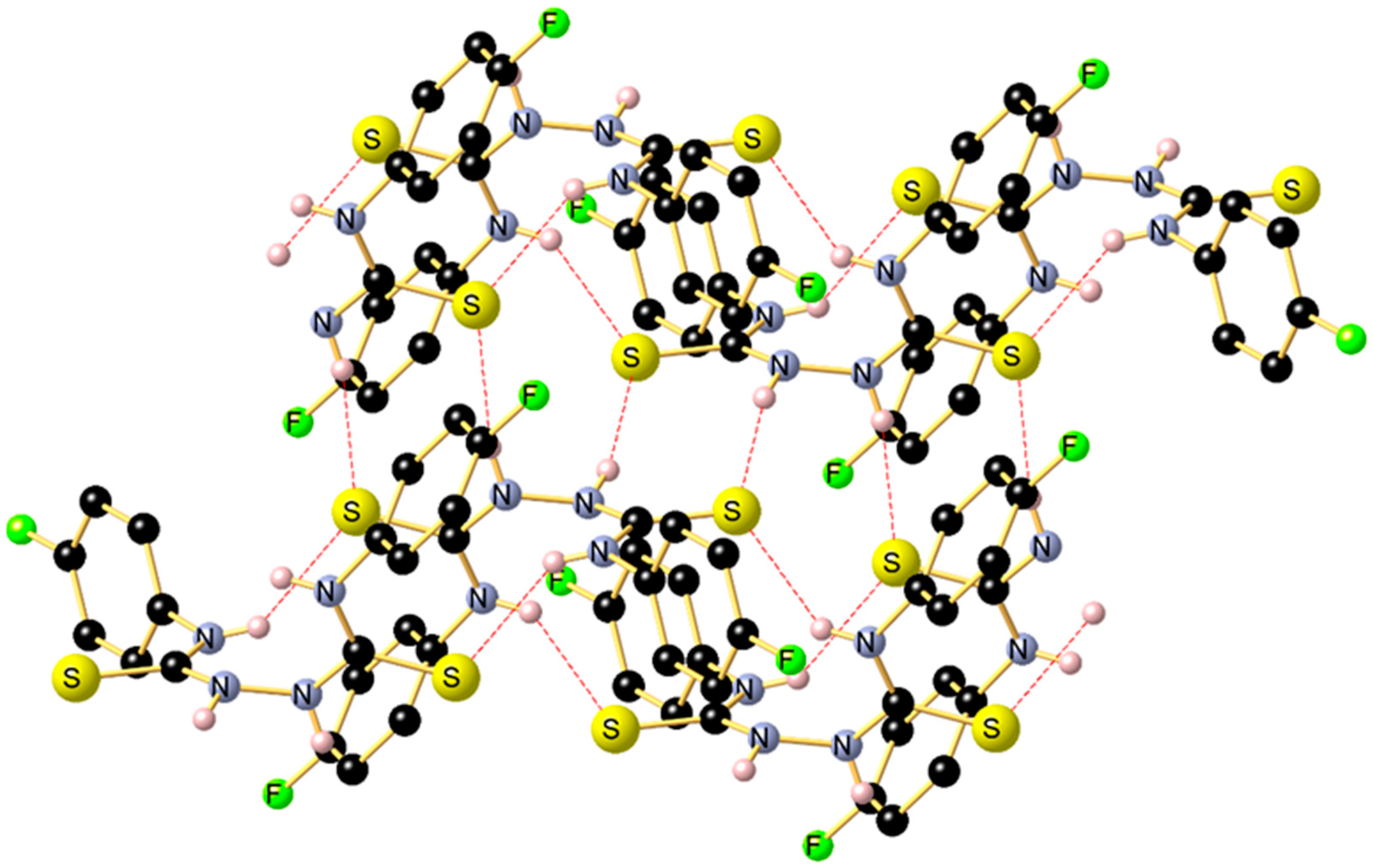
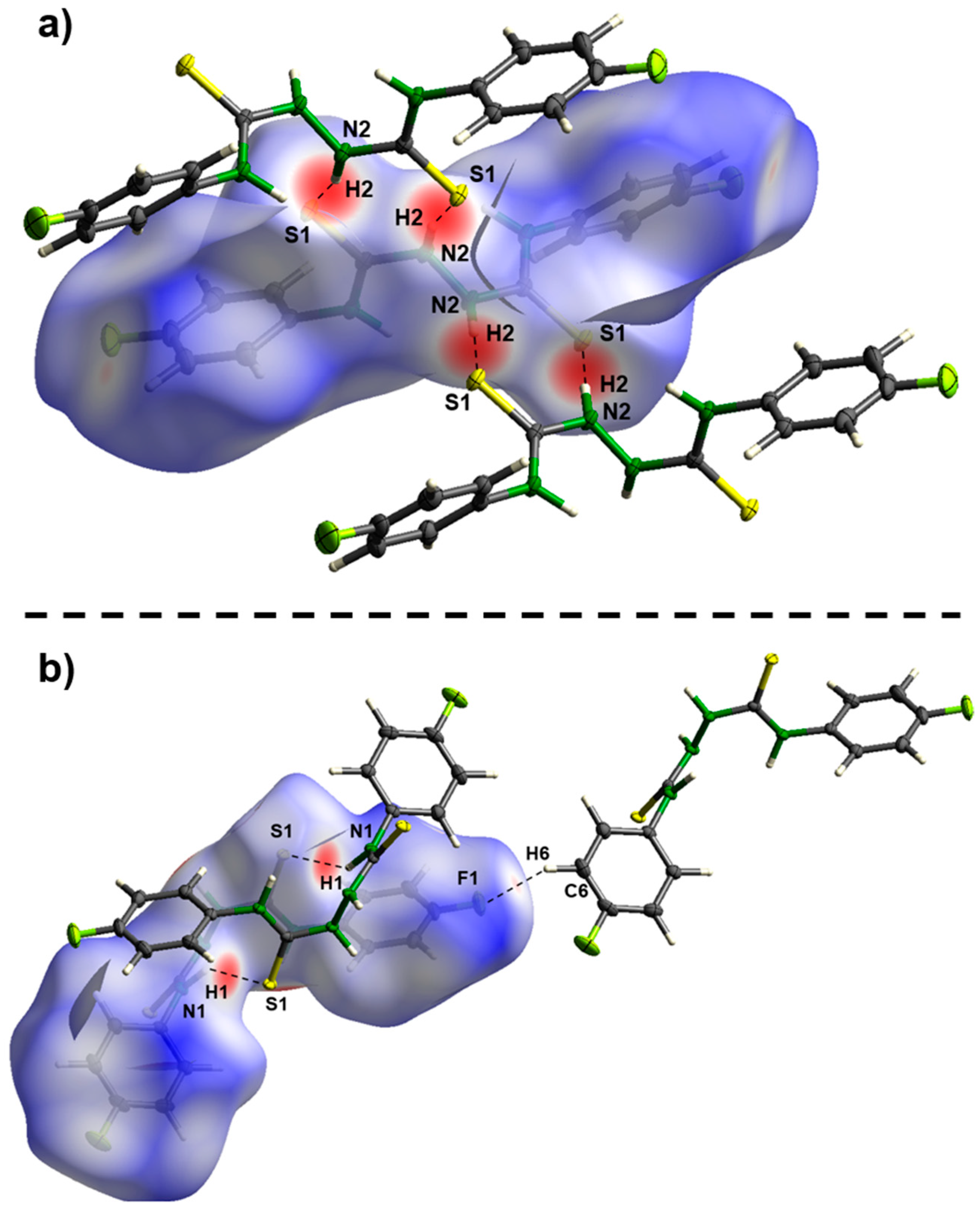
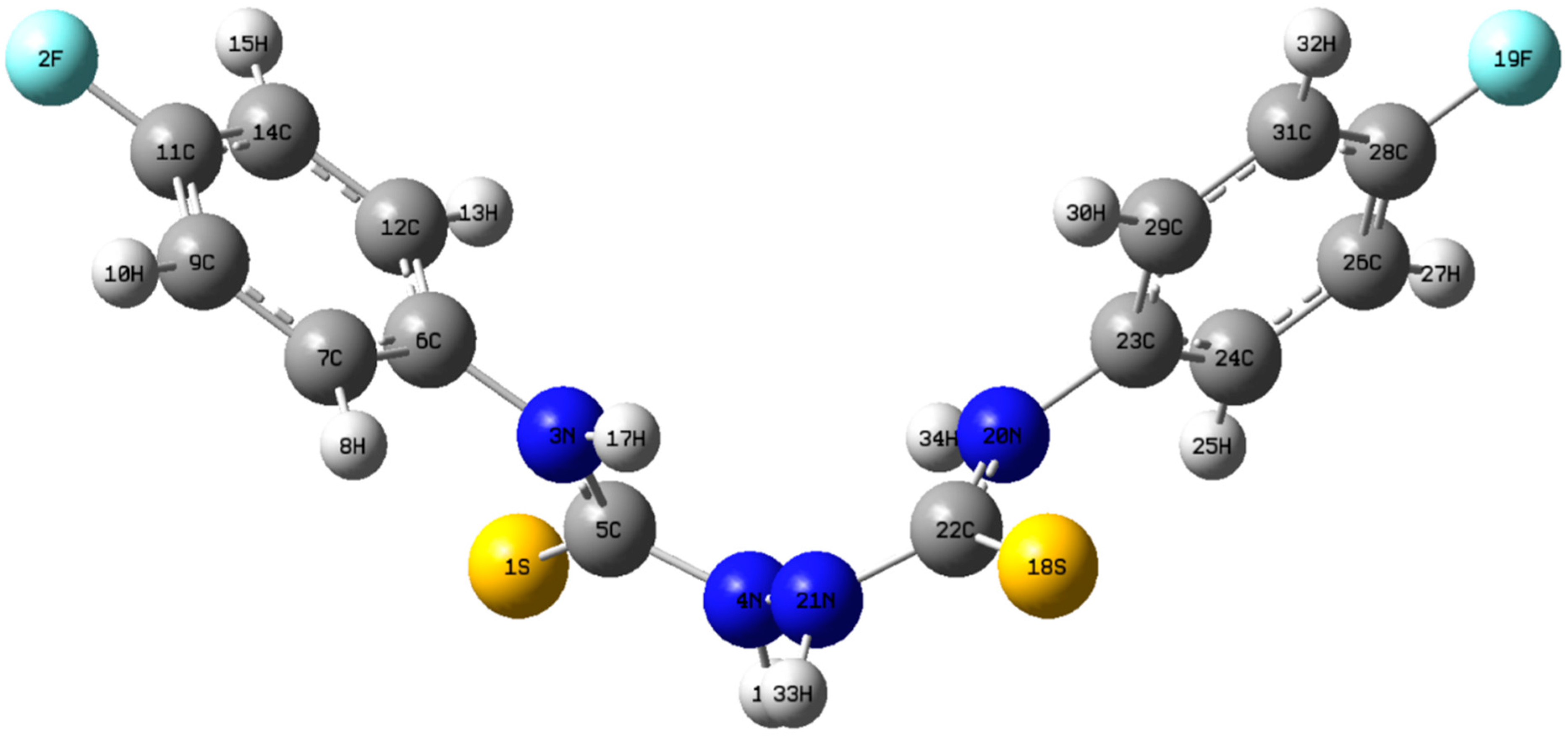
2. Results and Discussion
3. Materials and Methods
Theoretical Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kanzari-Mnallah, D.; Salhi, S.; Knorr, M.; Kirchhoff, J.L.; Strohmann, C.; Efrit, M.L.; Akacha, A.B. Synthesis of Isomeric β-Cycloalkoxyphosphonated Hydrazones Containing a Dioxaphosphorinane Ring: Conformational and Conformational Investigation and Molecular Docking Analysis. J. Mol. Struct. 2024, 1317, 139035. [Google Scholar] [CrossRef]
- Ben Akacha, A.; Barkallah, S.; Zantour, H. ¹³C NMR and ³¹P NMR Spectral Assignment of New β-Phosphonylated Hydrazones. Magn. Reson. Chem. 1999, 37, 916–920. [Google Scholar] [CrossRef]
- Kanzari-Mnallah, D.; Efrit, M.L.; Ben Akacha, A. Synthèse de 1,3,2-Dioxaphosphorinanes Diastéréoisomères: Influence de la Conformation des 1,3-Diols de Départ sur Leurs Structures et Conformations. Phosphorus Sulfur Silicon Relat. Elem. 2017, 192, 665–673. [Google Scholar] [CrossRef]
- Salah, N.; Arfaoui, Y.; Bahri, M.; Efrit, M.L.; Ben Akacha, A. Synthèse et Étude Conformationnelle par RMN (¹H, ¹³C, ³¹P) et DFT des Cycloalcoxyphosphinallenes et des Hydrazones β-Cycloalcoxyphosphonatées. Phosphorus Sulfur Silicon Relat. Elem. 2013, 188, 609–622. [Google Scholar] [CrossRef]
- Ben Akacha, A.; Ayed, N.; Baccar, B.; Charrier, C. Phospho-4-Pyrazoles. Synthesis and Proton, Phosphorus-31, and Carbon-13 NMR. Phosphorus Sulfur Relat. Elem. 1988, 40, 63–68. [Google Scholar] [CrossRef]
- Nurkenov, O.A.; Ibrayev, M.K.; Satpaeva Zh, B.; Dauletzhanova Zh, T.; Seilkhanov, T.M. Synthesis and Study of Antioxidant Activity of Hydrazone and Thiosemicarbazide Based on N-Morpholinoacetic Acid Hydrazide. Vestnik Karaganda Univ. Ser. Chem. 2018, 1, 22–26. [Google Scholar] [CrossRef]
- Gihsoyl, A.; Terzioglu, N.; Bik, G. Synthesis of Some New Hydrazide-Hydrazones, Thiosemicarbazides, and Thiazolidinones as Possible Antimicrobials. Eur. J. Med. Chem. 1997, 32, 753–757. [Google Scholar] [CrossRef]
- Salah, N.; Zribi, S.; Efrit, M.L.; Ben Akacha, A. Hydrazones β-Phosphonates: Nouvelles Voies d’Accès aux Thiosemicarbazones, 4-Phosphopyrazoles et Indoles 2-Phosphonates. J. Soc. Chim. Tunisie 2013, 15, 133–141. [Google Scholar]
- Ragab, A.; Ammar, Y.A.; Mahmoud, M.; Mohamed, B.I.; El-Tabl, S.; Abdou; Farag, S.S. Synthesis, Characterization, Thermal Properties, Antimicrobial Evaluation, ADMET Study, and Molecular Docking Simulation of New Mono Cu (II) and Zn (II) Complexes with 2-Oxoindole Derivatives. Comput. Biol. Med. 2022, 145, 105473. [Google Scholar] [CrossRef]
- Dharmasivam, M.; Kaya, B.; Wijesinghe, T.; Azad, M.G.; Gonzálvez, M.A.; Hussaini, M.; Chekmarev, J.; Bernhardt, P.V.; Richardson, D.R. Designing Tailored Thiosemicarbazones with Bespoke Properties: The Styrene Moiety Imparts Potent Activity, Inhibits Heme Center Oxidation, and Results in a Novel “Stealth Zinc (II) Complex”. J. Med. Chem. 2023, 66, 1426–1453. [Google Scholar] [CrossRef]
- Bal, T.R.; Anand, B.; Yogeeswari, P.; Sriram, D. Synthesis and Evaluation of Anti-HIV Activity of Isatin β-Thiosemicarbazone Derivatives. Bioorg. Med. Chem. Lett. 2005, 15, 4451–4455. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, R.B.; de Souza-Fagundes, E.M.; Soares, R.P.P.; Andrade, A.A.; Krettli, A.U.; Zani, C.L. Synthesis and Antimalarial Activity of Semicarbazone and Thiosemicarbazone Derivatives. Eur. J. Med. Chem. 2008, 43, 1983–1988. [Google Scholar] [CrossRef] [PubMed]
- Firdausiah, S.; Hasbullah, S.A.; Yamin, B.M. Synthesis, Structural Elucidation, and Antioxidant Study of Ortho-Substituted N, N′-bis(benzamidothiocarbonyl)hydrazine Derivatives. J. Phys. Conf. Ser. 2018, 979, 012010. [Google Scholar] [CrossRef]
- Mészáros Szécsényi, K.; Leovac, V.M.; Radosavljević Evans, I. Synthesis and Characterisation of a Novel Polymeric Cd Complex, Catena-(μ-Thio)[bis(N-Phenylthiourea)] bis(dimethylsulfoxide)dichlorocadmium (II). J. Coord. Chem. 2006, 59, 171–180. [Google Scholar] [CrossRef]
- Akinchan, N.T.; Drożdżewski, P.M.; Battaglia, L.P. Crystal structure and spectroscopic characterization of bis(N-phenylthiourea). J. Chem. Crystallogr. 2002, 32, 91–97. [Google Scholar] [CrossRef]
- Jaiswal, S.; Gond, M.K.; Bharty, M.K.; Maiti, B.; Krishnamoorthi, S.; Butcher, R.J. Manganese (II) catalyzed synthesis of bis (N-cyclohexylthiourea) derived from thiosemicarbazide: Structural characterization, fluorescence, cyclic voltammetry, Hirshfeld surface analysis and DFT calculation. J. Mol. Struct. 2021, 1246, 131060–131070. [Google Scholar] [CrossRef]
- Yamin, B.M.Y.; Yusof, M.S.M. N,N′-Bis(benzamidothiocarbonyl)hydrazine Dimethyl Sulfoxide Disolvate. Acta Crystallogr. Sect. E Struct. Rep. Online 2003, 59, o358–o359. [Google Scholar] [CrossRef]
- Deepthi, S.; Rajalakshmi, K.; Gunasekaran, K.; Velmurugan, D.; Nagarajan, K. Crystal and Molecular Structure of Addition Products of Dimethyl Acetylene Dicarboxylate and N,N′- Thiocarbanilyl Hydrazine. Mol. Cryst. Liq. Cryst. 2001, 369, 221–343. [Google Scholar] [CrossRef]
- Herrero, J.M.; Fabra, D.; Matesanz, A.I.; Hernández, C.; Sánchez-Pérez, I.; Quiroga, A.G. Dithiobiureas Palladium (II) Complexes’ Studies: From Their Synthesis to Their Biological Action. J. Inorg. Biochem. 2023, 246, 112261. [Google Scholar] [CrossRef]
- Spackman, P.R.; Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer: A Program for Hirshfeld Surface Analysis, Visualization, and Quantitative Analysis of Molecular Crystals. J. Appl. Cryst. 2021, 54, 1006–1011. [Google Scholar] [CrossRef]
- Bruker. Apex 4; Bruker AXS Inc.: Madison, WI, USA, 2021. [Google Scholar]
- Sheldrick, G.M. A Short History of SHELX. Acta Cryst. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. SHELXT—Integrated Space-Group and Crystal-Structure Determination. Acta Cryst. A 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Cryst. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A Complete Structure Solution, Refinement and Analysis Program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09: Revision A.1; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Dennington, R.D.; Keith, T.A.; Millam, J.M. GaussView 5.0.8; Gaussian Inc.: Wallingford, CT, USA, 2008. [Google Scholar]
- Becke, A.D. Density functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 1372–1377. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| D | H | A | dD–H/Å | dH–A/Å | dD–A/Å | D–H–A/° |
|---|---|---|---|---|---|---|
| N2 | H2 | S1 | 0.88(2) | 2.42(2) | 3.2749(16) | 164.4(19) |
| N1 | H1 | S1 | 0.85(2) | 2.56(2) | 3.2675(15) | 141.9(18) |
| C6 | H6 | F1 | 0.94(2) | 2.55(2) | 3.291(2) | 135.8(18) |
| C7 | H7 | F1 | 0.96(2) | 3.08(2) | 3.684(2 | 122.6(15) |
| Bond Lengths (Å) | Exp SCXRD | Calc. in EtOH ** | Calc. in MeOH | Calc. in MeCN | Calc. in CHCl3 | Angles (°) | Exp. SCXRD | Calc. in EtOH | Calc. in MeOH | Calc. in MeCN | Calc. in CHCl3 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| S1-C5 | 1.697(17) | 1.684 | 1.684 | 1.684 | 1.678 | C5-N4-N21 | 119.70(16) | 120.84 | 120.84 | 120.84 | 121.09 |
| F2-C11 | 1.366(2) | 1.361 | 1.361 | 1.361 | 1.358 | C5-N3-C6 | 122.29(14) | 127.48 | 127.46 | 127.45 | 127.61 |
| N4-N21 | 1.404(3) | 1.392 | 1.392 | 1.392 | 1.390 | N4-C5-S1 | 118.08(12) | 118.46 | 118.45 | 118.46 | 118.46 |
| N4-C5 | 1.367(2) | 1.388 | 1.387 | 1.387 | 1.390 | N3-C5-S1 | 123.81(13) | 126.85 | 126.81 | 126.81 | 127.11 |
| N3-C5 | 1.332(2) | 1.343 | 1.343 | 1.343 | 1.346 | C7-C6-N3 | 119.35(16) | 118.59 | 120.07 | 121.07 | 118.41 |
| N3-C6 | 1.440(2) | 1.426 | 1.426 | 1.426 | 1.427 | N3-C5-N4 | 118.08(15) | 114.68 | 114.71 | 114.72 | 114.38 |
| C6-C7 | 1.394(2) | 1.395 | 1.396 | 1.396 | 1.395 | F2-C11-C9 | 118.36(19) | 118.71 | 118.64 | 118.64 | 118.74 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salhi, S.; Kanzari-Mnallah, D.; Jourdain, I.; Knorr, M.; Strohmann, C.; Kirchhoff, J.-L.; Mrabet, H.; Ben Akacha, A. Synthesis of N-p-Fluorothiosemicarbazone and of Bis(N-p-Fluorophenylthiourea): Crystal Structure and Conformational Analysis of N,N′-Bis(4-Fluorophenyl)Hydrazine-1,2-Bis(Carbothioamide). Molbank 2024, 2024, M1926. https://doi.org/10.3390/M1926
Salhi S, Kanzari-Mnallah D, Jourdain I, Knorr M, Strohmann C, Kirchhoff J-L, Mrabet H, Ben Akacha A. Synthesis of N-p-Fluorothiosemicarbazone and of Bis(N-p-Fluorophenylthiourea): Crystal Structure and Conformational Analysis of N,N′-Bis(4-Fluorophenyl)Hydrazine-1,2-Bis(Carbothioamide). Molbank. 2024; 2024(4):M1926. https://doi.org/10.3390/M1926
Chicago/Turabian StyleSalhi, Sirine, Dorra Kanzari-Mnallah, Isabelle Jourdain, Michael Knorr, Carsten Strohmann, Jan-Lukas Kirchhoff, Hédi Mrabet, and Azaiez Ben Akacha. 2024. "Synthesis of N-p-Fluorothiosemicarbazone and of Bis(N-p-Fluorophenylthiourea): Crystal Structure and Conformational Analysis of N,N′-Bis(4-Fluorophenyl)Hydrazine-1,2-Bis(Carbothioamide)" Molbank 2024, no. 4: M1926. https://doi.org/10.3390/M1926
APA StyleSalhi, S., Kanzari-Mnallah, D., Jourdain, I., Knorr, M., Strohmann, C., Kirchhoff, J.-L., Mrabet, H., & Ben Akacha, A. (2024). Synthesis of N-p-Fluorothiosemicarbazone and of Bis(N-p-Fluorophenylthiourea): Crystal Structure and Conformational Analysis of N,N′-Bis(4-Fluorophenyl)Hydrazine-1,2-Bis(Carbothioamide). Molbank, 2024(4), M1926. https://doi.org/10.3390/M1926