
5-Diethoxymethyl-1,1-diethoxy-5-hydroxyundeca-3,6-diyn-2-one


{kind=link}
{kind=link}
Abstract
1. Introduction
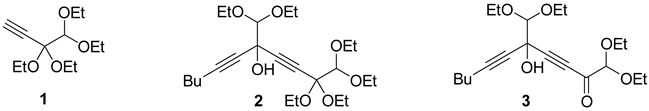
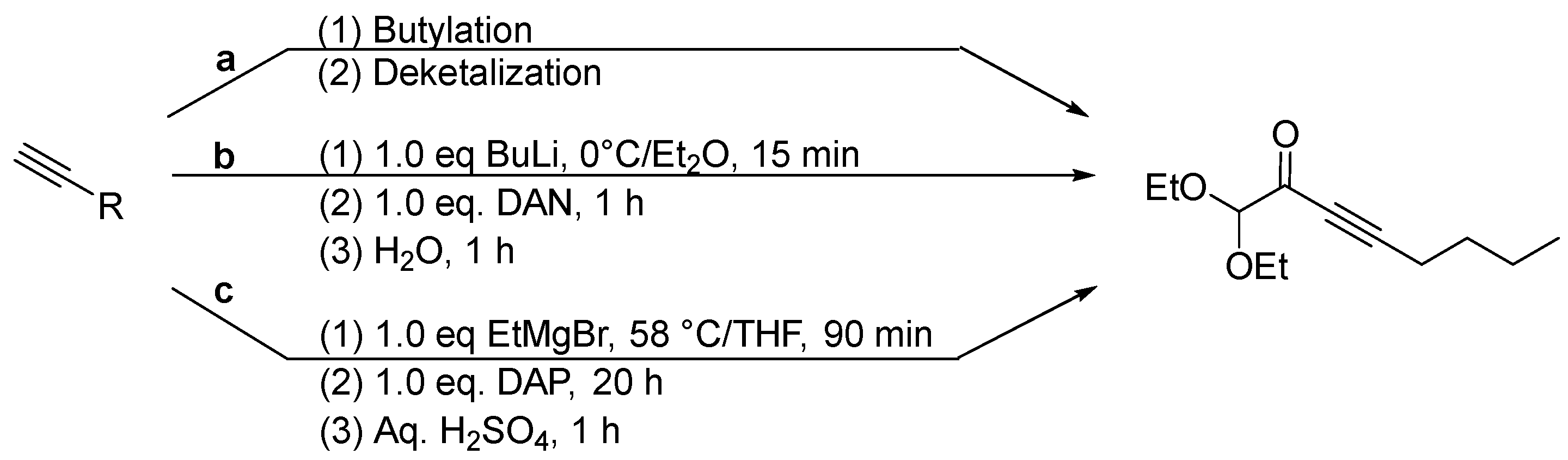
2. Results and Discussion
3. Conclusions
4. Materials and Methods
4.1. General Considerations
4.2. Preparation of 1,1-Diethoxyoct-3-yn-2-one
4.2.1. From 3,3,4,4-Tetraethoxybut-1-yne (1)
4.2.2. From 1-Hexyne by the Dulou Method
4.2.3. From 1-Hexyne by the Wohl–Lang Method
4.3. Preparation of 5-(Diethoxymethyl)-1,1,2,2-tetraethoxyundeca-3,6-diyn-5-ol (2)
4.4. Preparation of 5-(Diethoxymethyl)-1,1-diethoxy-5-hydroxyundeca-3,6-diyn-2-one (3)
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sydnes, L.K.; Holmelid, B.; Kvernenes, O.H.; Sandberg, M.; Hodne, M.; Bakstad, E. Synthesis and Some Chemical Properties of 3,3,4,4-Tetraethoxybut-1-yne. Tetrahedron 2007, 63, 4144–4148. [Google Scholar] [CrossRef]
- Valdersnes, S.; Sydnes, L.K. Preparation of 2-ethoxy-3-hydroxy-4-(perfluoroalkyl)tetrahydropyran derivatives from substituted 4-ethoxybut-3-en-1-ols. Eur. J. Org. Chem. 2009, 2009, 5816–5831. [Google Scholar] [CrossRef]
- Sydnes, L.K. Regiospecific preparation of substituted furans from some 5-substituted derivatives of 1,1-diethoxyalk-3-yn-2-ones. In Targets in Heterocyclic Systems; Chemistry and Properties; Attanasi, O.A., Merino, P., Spinelli, D., Eds.; Italian Society of Chemistry: Rome, Italy, 2016; Volume 20, pp. 316–336. [Google Scholar] [CrossRef]
- Tian, S.K.; Hong, R.; Deng, L. Catalytic Asymmetric Cyanosilylation of Ketones with Chiral Lewis Base. J. Am. Chem. Soc. 2003, 125, 9900–9901. [Google Scholar] [CrossRef] [PubMed]
- Dulou, R.; Savostianoff, D. Synthèse d’acétals de Glyoxals à Partir du Diéthoxyacétonitrile. C. R. Hebd. Seances Acad. Sci. Série C Sci. Chim. 1966, 252, 564–566. [Google Scholar]
- Wohl, A.; Lange, M. Aufbau Des Milchsäurealdehyds. Berichte Dtsch. Chem. Ges. 1908, 41, 3612–3620. [Google Scholar] [CrossRef]
- Serratosa, F. An Acetylenic Approach to Patulin Derivatives. Tetrahedron 1961, 16, 185–191. [Google Scholar] [CrossRef]
- Williams, D.B.G.; Cullen, A.; Fourie, A.; Henning, H.; Lawton, M.; Mommsen, W.; Nangu, P.; Parker, J.; Renison, A. Mild Water-Promoted Selective Deacetalisatison of Acyclic Acetals. Green Chem. 2010, 12, 1919. [Google Scholar] [CrossRef]
- De Kimpe, N.; Stevens, C. Synthesis of the principal bread flavor component, 6-acetyl-1,2,3,4-tetrahydropyridine, and acetal protected precursors. Tetrahedron 1995, 51, 2387–2402. [Google Scholar] [CrossRef]
- Huet, F.; Lechevallier, A.; Pellet, M.; Conia, J.M. Wet silica gel; a convenient reagent for deacetalization. Synthesis 1978, 1978, 63–65. [Google Scholar] [CrossRef]
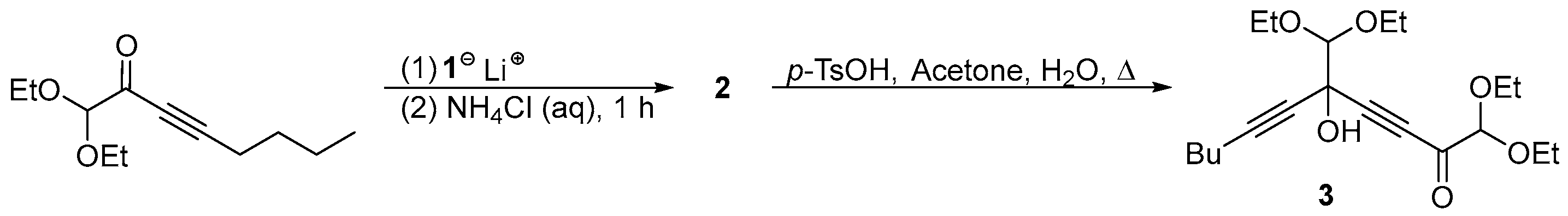
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Espeland, L.O.; Sydnes, L.K. 5-Diethoxymethyl-1,1-diethoxy-5-hydroxyundeca-3,6-diyn-2-one. Molbank 2024, 2024, M1896. https://doi.org/10.3390/M1896
Espeland LO, Sydnes LK. 5-Diethoxymethyl-1,1-diethoxy-5-hydroxyundeca-3,6-diyn-2-one. Molbank. 2024; 2024(4):M1896. https://doi.org/10.3390/M1896
Chicago/Turabian StyleEspeland, Ludvik O., and Leiv K. Sydnes. 2024. "5-Diethoxymethyl-1,1-diethoxy-5-hydroxyundeca-3,6-diyn-2-one" Molbank 2024, no. 4: M1896. https://doi.org/10.3390/M1896
APA StyleEspeland, L. O., & Sydnes, L. K. (2024). 5-Diethoxymethyl-1,1-diethoxy-5-hydroxyundeca-3,6-diyn-2-one. Molbank, 2024(4), M1896. https://doi.org/10.3390/M1896