
Thiazolylketol Acetates as Glycosyl Donors: Stereoselective Synthesis of a C-Ketoside



Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
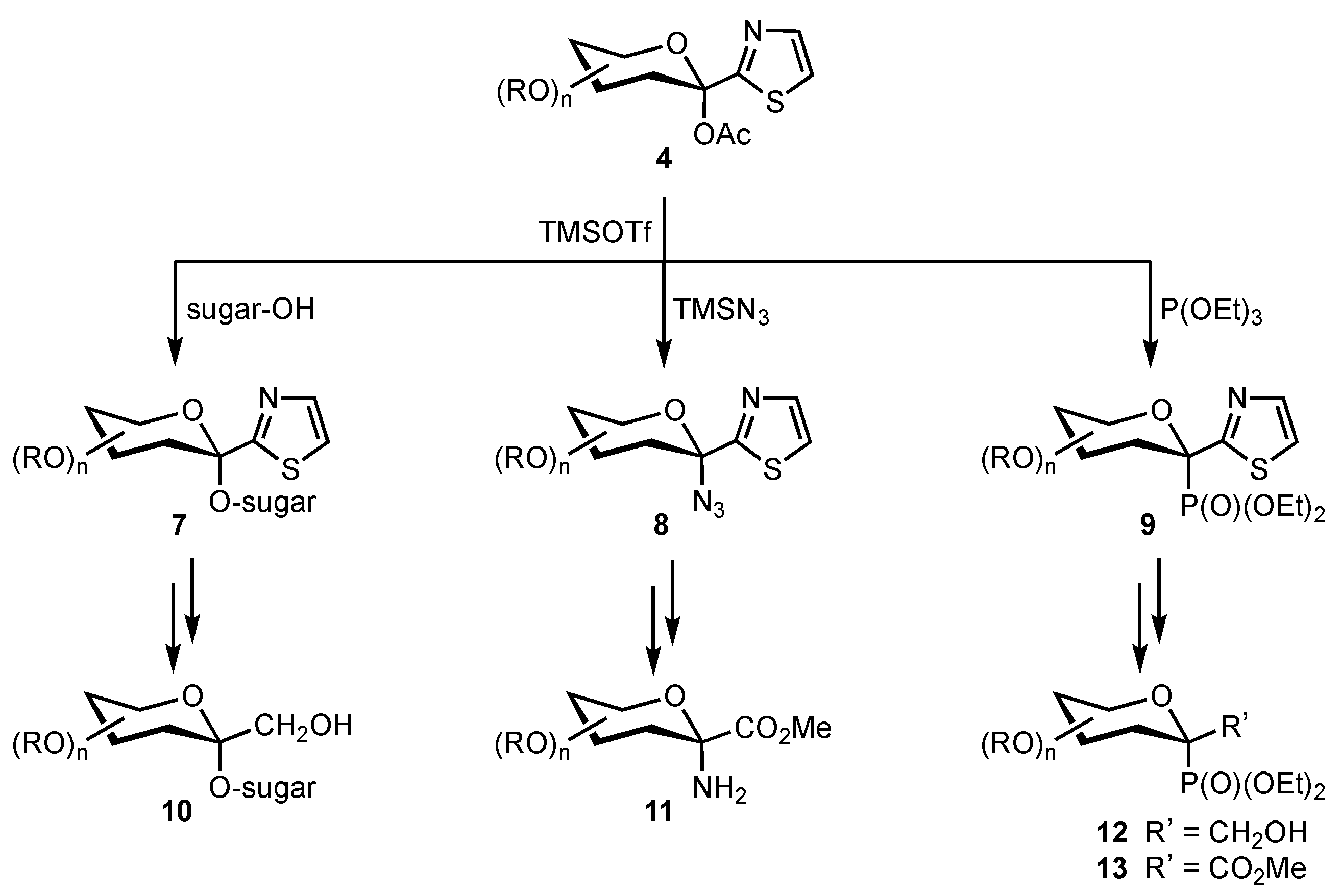
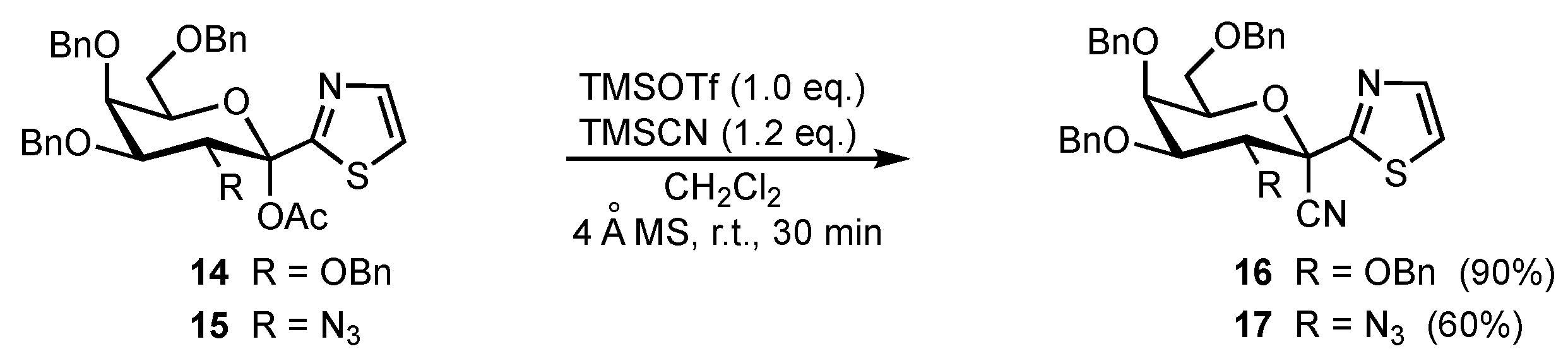
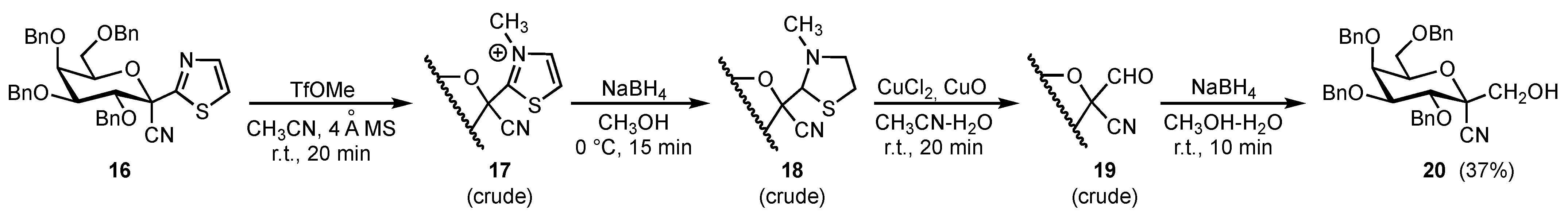
2. Results and Discussion
3. Materials and Methods
3.1. (1R)-2,3,4,6-Tetra-O-benzyl-1-C-cyano-1-deoxy-1-C-(2-thiazolyl)-D-galactopyranose (16)
3.2. (1S)-2-Azido-3,4,6-tri-O-benzyl-1-C-cyano-1-deoxy-1-C-(2-thiazolyl)-D-galactopyranose (17)
3.3. 2,6-Anhydro-3,4,5,7-tetra-O-benzyl-2-C-cyano-D-glycero-L-manno-heptitol (20)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Postema, M.H.D. Recent developments in the synthesis of C-glycosides. Tetrahedron 1992, 48, 8545–8599. [Google Scholar] [CrossRef]
- Postema, M.H.D. C-glycoside Synthesis; CRC Press: Boca Ratón, FL, USA, 1995. [Google Scholar] [CrossRef]
- Levy, D.E.; Tang, C. The Chemistry of C-Glycosides; Pergamon Press: Oxford, UK, 1995. [Google Scholar]
- Wellington, K.W.; Benner, S.A. A Review: Synthesis of Aryl C-Glycosides via the Heck Coupling Reaction. Nucleosides Nucleotides Nucleic Acids 2006, 25, 1309–1333. [Google Scholar] [CrossRef] [PubMed]
- Lalitha, K.; Muthusamy, K.; Prasad, Y.S.; Vemula, P.K.; Nagarajan, S. Recent developments in β-C-glycosides: Synthesis and applications. Carbohydr. Res. 2015, 402, 158–171. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Yu, B. Recent Advances in the Chemical Synthesis of C-Glycosides. Chem. Rev. 2017, 117, 12281–12356. [Google Scholar] [CrossRef] [PubMed]
- Liao, H.; Ma, J.; Yaoa, H.; Liu, X.-W. Recent progress of C-glycosylation methods in the total synthesis of natural products and pharmaceuticals. Org. Biomol. Chem. 2018, 16, 1791–1806. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, K.; Ando, Y.; Matsumoto, T.; Suzuki, K. Total Synthesis of Aryl C-Glycoside Natural Products: Strategies and Tactics. Chem. Rev. 2018, 118, 1495–1598. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.-Y.; Fan, N.-L.; Hu, X.-G. Recent development in the synthesis of C-glycosides involving glycosyl radicals. Org. Biomol. Chem. 2020, 18, 5095–5109. [Google Scholar] [CrossRef] [PubMed]
- Gosh, T.; Nokami, T. Recent development of stereoselective C-glycosylation via generation of glycosyl radical. Carbohydr. Res. 2022, 522, 108677. [Google Scholar] [CrossRef] [PubMed]
- Shang, W.; Niu, D. Radical Pathway Glycosylation Empowered by Bench-Stable Glycosyl Donors. Acc. Chem. Res. 2023, 56, 2473–2488. [Google Scholar] [CrossRef] [PubMed]
- Shang, W.; Shi, R.; Niu, D. C-Glycoside Synthesis Enabled by Nickel Catalysis. Chin. J. Chem. 2023, 41, 2217–2236. [Google Scholar] [CrossRef]
- Gou, X.-Y.; Zhu, X.-Y.; Zhang, B.-S.; Liang, Y.-M. Synthesis of C-Aryl Glycosides by C−H Functionalization. Chem. Eur. J. 2023, 29, e202203351. [Google Scholar] [CrossRef] [PubMed]
- Achanta, S.; Sasikala, C.V.A.; Basu, D.; Nahide, P.D.; Bandichhor, R. Stereochemical Aspects of the C-Glycosylation of Pyranosides and Furanosides. Synthesis 2024, 56, 1043–1069. [Google Scholar] [CrossRef]
- Das, R.; Das, M.; Mukherjee, D.; Kundu, T. Recent Advances on the Synthesis of C-Glycosides from 1,2-Glycals. Synthesis 2024, 56, 1070–1096. [Google Scholar] [CrossRef]
- Dondoni, A.; Scherrmann, M.-C. Thiazole-Based Synthesis of Formyl C-Glycosides. J. Org. Chem. 1994, 59, 6404–6412. [Google Scholar] [CrossRef]
- Dondoni, A.; Marra, A. Thiazolylketoses: A new class of versatile intermediates for glycoside synthesis. Chem. Commun. 1999, 2133–2145. [Google Scholar] [CrossRef]
- Haverkamp, J.; Spoormaker, T.; Dorland, L.; Vliegenthart, J.F.G.; Schauer, R. Determination of the β-Anomeric Configuration of Cytidine 5′-Monophospho-N-acetylneuraminic Acid by 13C NMR Spectroscopy. J. Am. Chem. Soc. 1979, 101, 4851–4853. [Google Scholar] [CrossRef]
- Hori, H.; Nakajima, T.; Nishida, Y.; Ohrui, H.; Meguro, H. A simple method to determine the anomeric configuration of sialic acid and its derivatives by 13C-NMR. Tetrahedron Lett. 1988, 29, 6317–6320. [Google Scholar] [CrossRef]
- Pritulla, S.; Lauterwein, J.; Klessinger, M.; Thiem, J. Configurational assignment of N-acetylneuraminic acid and analogues via the vicinal C,H coupling constants. Carbohydr. Res. 1991, 215, 345–349. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferrari, C.; Dondoni, A.; Marra, A. Thiazolylketol Acetates as Glycosyl Donors: Stereoselective Synthesis of a C-Ketoside. Molbank 2024, 2024, M1883. https://doi.org/10.3390/M1883
Ferrari C, Dondoni A, Marra A. Thiazolylketol Acetates as Glycosyl Donors: Stereoselective Synthesis of a C-Ketoside. Molbank. 2024; 2024(3):M1883. https://doi.org/10.3390/M1883
Chicago/Turabian StyleFerrari, Clark, Alessandro Dondoni, and Alberto Marra. 2024. "Thiazolylketol Acetates as Glycosyl Donors: Stereoselective Synthesis of a C-Ketoside" Molbank 2024, no. 3: M1883. https://doi.org/10.3390/M1883
APA StyleFerrari, C., Dondoni, A., & Marra, A. (2024). Thiazolylketol Acetates as Glycosyl Donors: Stereoselective Synthesis of a C-Ketoside. Molbank, 2024(3), M1883. https://doi.org/10.3390/M1883