1. Introduction
Quinones represent a class of biologically significant compounds endowed with a complex and fascinating chemistry [
1,
2]. They are widely distributed in nature, being found in bacteria, plants, arthropods, marine invertebrates, and mammals [
3,
4,
5], and play crucial roles in several cell signaling pathways. Indeed, they are involved in primary metabolic processes such as photosynthesis and respiration, acting as electron transfer agents [
6]; they actively participate in biological redox processes, especially. Under physiological conditions, they can undergo a non-enzymatic one-electron reduction to produce the toxic species of semiquinone radical-anion which determine their activities, e.g., toxicities in the cellular systems [
7,
8].
The basic chemical motif of quinone-derived compounds is 1,4-benzoquinone, which exhibits prominent biological and pharmacological applications [
3]. Striking examples are the ubiquinones, involved in the human respiratory chain and acting as essential electron-transfer agents, or doxorubicin, one of the first-line agents in cancer chemotherapy treatment [
6]. Furthermore, 1,4-benzoquinone derivatives are key intermediates in the biosynthesis of antibiotics such as tetracyclines, although most studies on quinones are related to their cytotoxic effects, which are closely related to their ability to undergo redox cycling processes with the production of toxic reactive oxygen species (ROSs) [
7,
9]. More generally, the most recent literature shows that quinones have huge potential for pharmacological applications, e.g., as an anti-inflammatory, immunomodulatory, cardioprotective, antidiabetic, neuroprotective, antiepileptic, antibacterial, antifungal, antiparasitic, and antiviral [
9,
10,
11,
12,
13]. As a result of this, quinones have also become of great interest in the synthetic organic chemistry field and the 1,4-benzoquinone, in particular, represents a valuable building block and serves also as key chemical motif in the synthetic generation of many biologically active compounds. Currently, the diversity-oriented synthesis (DOS) approach based on this chemical scaffold is an extremely active area of research worldwide, with an increasing demand for optimized synthetic procedures.
The main synthetic approach to obtain 1,4-benzoquinones consists of the treatment of 1,4-dimethoxybenzene derivatives with oxidizing agents, mostly ceric ammonium nitrate (CAN) [
14,
15]. The treatment of hydroquinone ethers with CAN is also the most widely used protocol for the synthesis of diquinones [
16,
17], which also represent a group of chemically and pharmacologically interesting compounds. For instance, they serve as chemical starting point for the synthesis of popolohuanone E, a topoisomerase II inhibitor [
18]; moreover, the diquinone system is found in an interesting group of biologically active natural products which have been found in many plants [
19,
20,
21].
Within the context of our search for novel antiparasitic agents, we have recently created a chemical library of quinone derivatives in a DOS campaign, where the synthetic structures were based on natural bioactive metabolites of marine origin [
22]. We have rationally designed and prepared a set of quinone-derived compounds for SAR studies and mechanism of action studies. Indeed, we have demonstrated their ability to undergo one-electron reduction, resulting in a toxic semiquinone radical species able to impair parasites viability [
23,
24].
In this DOS campaign, we paid special attention to the oxidation step and to the feasibility of modulating the ratio quinone/diquinone in the reaction product. Herein, we report the synthesis of 2-(4-benzyl substituted)-1,4-dimethoxybenzene derivatives by evaluating the effects of a different mode in which CAN is added to the arene solution for the generation of quinones. In this study, we observed a different quinone/diquinone product ratio depending on a “traditional” or “inverse” addition of the oxidizing agent consistent with the findings of Love and coworkers [
15].
2. Results and Discussion
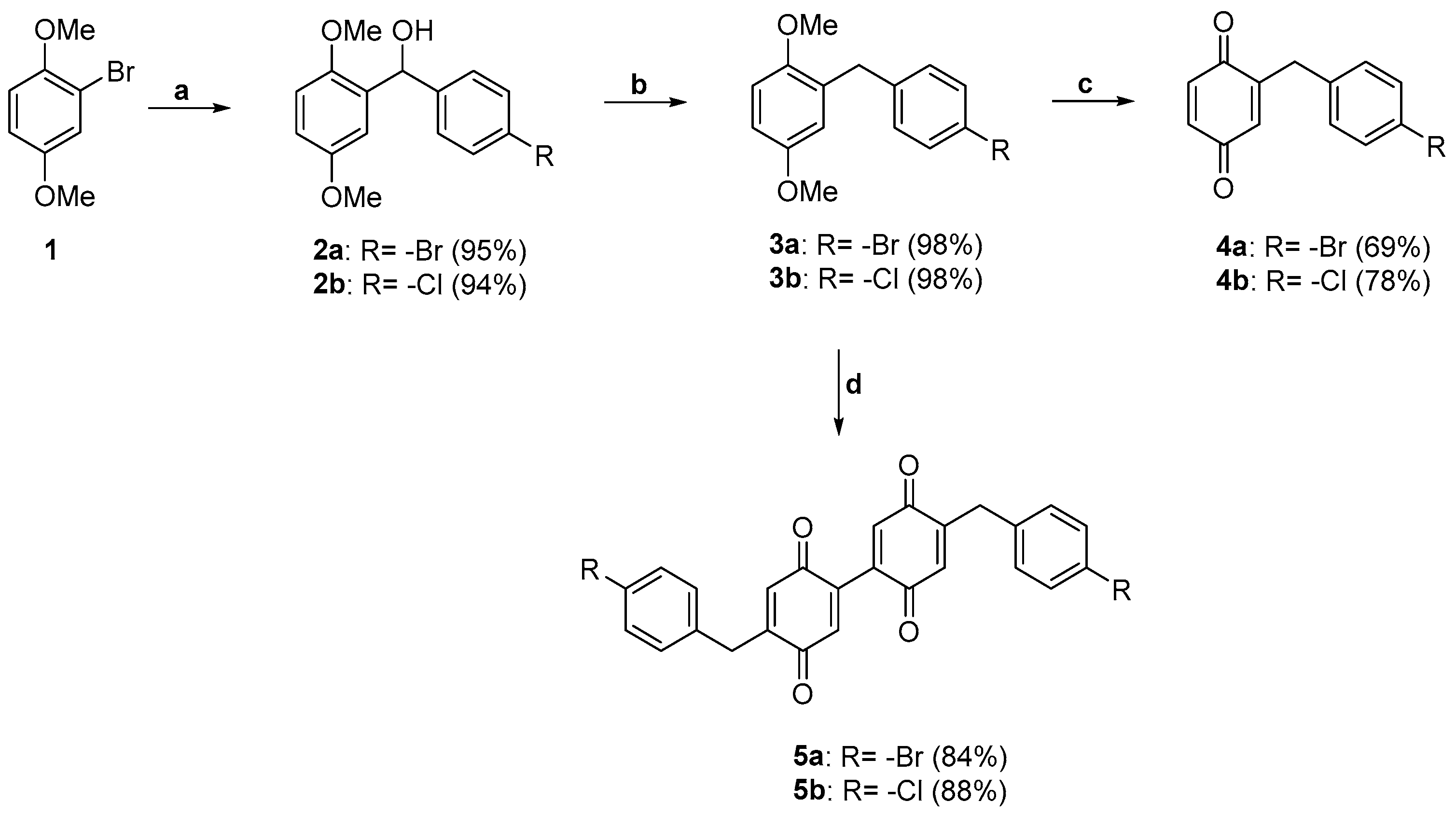
With the aim to easily enlarge the chemical diversity of the quinone-based compounds library, we planned to prepare quinones (
4a and
4b) and diquinones (
5a and
5b) from the same 2-(4-benzyl-substituted)-1,4-dimethoxybenzene starting material. As already reported in the literature, indeed, the manner by which the oxidation step with ceric ammonium nitrate (CAN) is carried out strongly affects the quinone/diquinone product ratios [
15,
16,
23]. Therefore, in this study, we adopted different reaction protocols to enable the selective synthesis of quinones [
23] or the corresponding diquinones [
15], observing the effects of CAN oxidation when the 4-bromo- and 4-chlorobenzyl groups are bound to the 1,4-dimethoxybenzene moiety.
The commercially available 1-bromo-2,5-dimethoxybenzene (
1) was first converted into its lithiated derivative with n-butyllithium in THF solution, and then coupled with a suitable aldehyde to afford the benzyl alcohols
2a and
2b (
Scheme 1). A large excess of trifluoroacetic acid was added to a dichloromethane solution of the latter, followed by treatment with triethylsilane (TES), which acts as a source of hydride ions. This method led to the benzyl derivatives
3a and
3b in pure form by a quantitative reaction.
Our previously developed method of 1,2,4-trimethoxybenzene derivatives’ oxidation with CAN [
23,
24] was applied to these substrates, following a procedure called “traditional” addition. In parallel, we performed the same oxidation reaction, slightly modifying the mode by which CAN is added, adopting the so-called “inverse” addition [
15].
Table 1 shows the procedure details, the isolated yield, and the quinone/diquinone product formation. We successfully demonstrated that when the arene substrates
3a and
3b (entries 1 and 2,
Table 1) were dissolved in acetonitrile and the aqueous solution CAN is directly and slowly added (traditional mode), the quinone formation is favored. Indeed, quinones
4a and
4b are the only observed products of this reaction with a higher percentage yield when the
para group of the benzyl substituent is a chlorine atom (69% for
4a vs. 78% for
4b). On the other hand, the inverse addition of CAN converted the arenes
3a and
3b mainly into the relevant diquinones
5a and
5b, with very high yields (entries 3 and 4,
Table 1). In these cases, a 2 mmol/mL aqueous solution of the oxidizing agent CAN is prepared, and to it the acetonitrile solution of compounds
3a and
3b (1 mmol/mL) is added. This latter outcome can be interpreted by taking into account that shifting the CH
3CN/H
2O ratio in the reaction solution from 75:25 (traditional addition) to 35:65 (inverse addition) drives the formation of aggregates of the arene molecules and, thus, strongly addresses toward the diquinone formation for such halogenated substrates (
3a and
3b) due to their poor solubility in water. This aggregation would be not expected to occur when traditional protocol is applied, since aqueous CAN is added dropwise to the acetonitrile solution of arenes. This allows the percentage of water to be kept low, thus increasing the solubility of arenes in the mixture of the reaction.
3. Materials and Methods
3.1. Materials
Reagents and solvents were purchased from Sigma-Aldrich (Saint Louis, MO, USA) and used without further purification. TLC (Silica Gel 60 F254, plates 5 × 20, 0.25 mm) were purchased from Merck (Kenilworth, NJ, USA). High-resolution electrospray ionization–mass spectrometry (ESI-MS) was performed on a Thermo LTQ Orbitrap XL spectrometer (Thermo-Fisher, San Josè, CA, USA). The MS spectra were recorded by infusion into the ESI source using methanol as solvent. The
1H (600 MHz) and
13C (175 MHz) NMR experiments were carried out on a Bruker Avance Neo spectrometer (Bruker BioSpin Corporation, Billerica, MA, USA); chemical shifts were reported in parts per million (ppm) and referenced to the residual solvent signal (CHCl
3:
δH = 7.26;
δC = 77.0). The monodimensional
1H NMR spectra were transformed at 64 K points with a digital resolution of 0.09 Hz for an accurate measurement of the coupling constants [
25]. High-performance liquid chromatography (HPLC) separation was achieved on Knauer Azura Pump 4.1 S instrument equipped with a Knauer K-2301 RI detector (Lab-Service Analytica s.r.l., Anzola dell’Emilia, Italy).
3.2. Synthesis of Compounds 2a and 2b
Both derivatives 2a and 2b were synthesized from the commercially available 1-bromo-2,5-dimethoxybenzene (280.0 µL, 1.84 mmol) that was dissolved in dry THF (5.0 mL) and stirred under an Argon atmosphere in ice bath. To each mixture, n-BuLi (1.70 mL, 2.95 mmol, 1.6 equiv.) was slowly added dropwise over 10 min, stirring the mixture for 1 h at 0 °C. After this time, a THF solution (1.0 mL) of a properly aldehyde [4-bromobenzaldehyde (500.0 mg, 2.76 mmol, 1.5 equiv.) or 4-chlorobenzaldehyde (380.0 mg, 2.76 mmol, 1.5 equiv.] was added, warming the resultant mixture at room temperature (rt). After being stirred for 6 h, the reaction was quenched with an aqueous solution of saturated NH4Cl and extracted twice with Et2O (2 × 100 mL). The combined organic layers were washed with brine, dried over Na2SO4, filtered, and concentrated under vacuum. In both cases, the crude material was chromatographed by HPLC on SiO2 (Kromasil SiO2 10 µM column, hexane/EtOAc 7:3 v/v%, flow rate 3.0 mL/min) providing compound 2a (tR = 10.1 min, 569.1 mg, 95%) and compound 2b (tR = 20.3 min, 486.2, 94%) in pure form.
Compound 2a, (4-bromophenyl)(2,5-dimethoxyphenyl)methanol: yellowish oil. 1H NMR (600 MHz) in CDCl3: δH = 7.46 (2H, d, J = 8.5 Hz), 7.28 (2H, d, J = 8.5 Hz), 6.85 (1H, d, J = 2.7 Hz), 6.82–6.79 (2H, overlapped), 5.96 (1H, s), 3.77 (6H, s). 13C NMR (175 MHz) in CDCl3: δC = 153.8 (C), 150.8 (C), 142.3 (C), 132.6 (C), 131.3 (CH), 128.3 (CH), 121.1 (C), 114.0 (CH), 112.9 (CH), 111.9 (CH), 71.6 (CH), 55.9 (CH3), 55.8 (CH3). HRESIMS m/z 345.0085 [M + Na]+ (calcd. 345.0097 for C15H15O3BrNa).
Compound 2b, (4-chlorophenyl)(2,5-dimethoxyphenyl)methanol: yellowish oil. 1H NMR (600 MHz) in CDCl3: δH = 7.18 (2H, d, J = 8.7 Hz), 7.14 (2H, d, J = 8.7 Hz), 6.74 (1H, d, J = 2.8 Hz), 6.67 (1H, d, J = 8.8 Hz), 6.65 (1H, dd, J = 8.8, 2.8 Hz), 5.83 (1H, s), 3.60 (6H, s). 13C NMR (175 MHz) in CDCl3: δC = 153.8 (C), 150.7 (C), 141.8 (C), 132.8 (C), 132.7 (CH), 128.3 (CH), 127.9 (CH), 113.8 (C), 112.9 (CH), 111.8 (CH), 71.3 (CH), 55.9 (CH3), 55.7 (CH3). HRESIMS m/z 301.0593 [M + Na]+ (calcd. 301.0602 for C15H15O3ClNa).
3.3. Synthesis of Compounds 3a and 3b
To an aliquot of compound 2a (225.0 mg, 0.698 mmol) or compound 2b (191.0 mg, 0.685 mmol) in CH2Cl2 (10.0 mL), an excess of trifluoroacetic acid was added dropwise and stirred for 10 min at room temperature (TFA 400.0 μL, 5.22 mmol). After this time, triethylsilane (TES, 340.0 μL, 2.1 mmol, 3 equiv.) was added, and the reaction was stirred overnight under vigorous magnetic stirring at room temperature. Then, the solvent was evaporated under vacuum, and the residue was diluted with an aqueous saturated solution of NaHCO3 (50 mL) and extracted with CH2Cl2 (2 × 50 mL). The organic phases were washed with brine, dried over Na2SO4, and filtered and concentrated under vacuum-affording compounds 3a and 3b in enough pure form.
Compound 3a, 2-(4-bromobenzyl)-1,4-dimethoxybenzene: yellowish oil (210.2 mg, 98%). 1H NMR (600 MHz) in CDCl3: δH = 7.23 (2H, d, J = 8.5 Hz), 6.93 (2H, d, J = 8.5 Hz), 6.64 (1H, d, J = 8.9 Hz), 6.58 (1H, dd, J = 8.9, 2.2 Hz), 6.51 (1H, d, J = 2.2 Hz), 3.75 (2H, s), 3.61 (3H, s), 3.58 (3H, s). 13C NMR (175 MHz) in CDCl3: δC = 153.8 (C), 151.6 (C), 139.8 (C), 131.3 (CH), 130.7 (CH), 130.2 (C), 119.7 (C), 116.8 (CH), 111.4 (CH), 55.9 (CH3), 55.7 (CH3), 35.6 (CH2). HRESIMS m/z 307.0334 [M + H]+ (calcd. 307.0328 for C15H16O2Br).
Compound 3b, 2-(4-chlorobenzyl)-1,4-dimethoxybenzene: yellowish oil (176.4 mg, 98%). 1H NMR (600 MHz) in CDCl3: δH = 7.23 (2H, d, J = 8.3 Hz), 7.14 (2H, d, J = 8.3 Hz), 6.80 (1H, d, J = 9.6 Hz), 6.74 (1H, dd, J = 9.6, 2.6 Hz), 6.65 (1H, d, J = 2.6 Hz), 3.91 (2H, s), 3.77 (3H, s), 3.73 (3H, s). 13C NMR (175 MHz) in CDCl3: δC = 153.5 (C), 151.6 (C), 139.3 (C), 131.6 (C), 130.3 (CH), 128.4 (CH), 116.8 (CH), 111.5 (CH), 111.4 (CH), 55.9 (CH3), 55.7 (CH3), 35.5 (CH2). HRESIMS m/z 285.0647 [M + Na]+ (calcd. 285.0653 for C15H15O2ClNa).
3.4. Synthesis of Compounds 4a and 4b
Procedure A: The synthesis of quinones
4a and
4b was performed following our previously developed procedure [
23,
24]. Compound
3a (65.8 mg, 0.22 mmol) and compound
3b (57.6 mg, 0.22 mmol) were separately dissolved in acetonitrile (12.0 mL) and cooled at 0 °C in an ice-bath. For each oxidation, a water solution (4 mL) of ceric ammonium nitrate (1 equiv. of CAN/mL) was slowly added over 15 min, observing a rapid mixture toning from yellow to orange. The reaction was constantly monitored by TLC (hexane/EtOAc 95:5
v/
v%), and it was quenched when the starting material’s spot appeared constantly weak (45 min). The reaction was firstly warmed at room temperature, diluted with H
2O (60 mL), and extracted with Et
2O (2 × 50 mL). The combined organic layer was washed with brine, dried, filtered and concentrated under vacuum. Both raw materials were purified by HPLC, providing the quinones
4a and
4b in pure state.
Compound 4a, 2-(4-bromobenzyl)cyclohexa-2,5-diene-1,4-dione: the crude material was chromatographed by HPLC on SiO2 (Kromasil SiO2 10 µM column, hexane/EtOAc 9:1 v/v%, flow rate 3.0 mL/min) giving the quinone 4a in pure form (yellowish solid, tR = 18.1 min, 69%). 1H NMR (600 MHz) in CDCl3: δH = 7.44 (2H, d, J = 8.1 Hz), 7.07 (2H, d, J = 8.1 Hz), 6.77 (1H, d, J = 10.0 Hz), 6.71 (1H, dd, J = 10.0, 2.5 Hz), 6.37 (1H, s), 3.69 (2H, s). 13C NMR (175 MHz) in CDCl3: δC = 187.4 (CO), 186.9 (CO), 148.0 (C), 136.5 (CH), 136.3 (CH), 134.7 (C), 133.2 (CH), 132.8 (C), 130.5 (CH), 128.8 (CH), 34.5 (CH2). HRESIMS m/z 298.9678 [M + Na]+ (calcd. 298.9677 for C13H9O2BrNa).
Compound 4b, 2-(4-chlorobenzyl)cyclohexa-2,5-diene-1,4-dione: the crude material was chromatographed by RP-HPLC (Luna 5 µM C18 column, CH3CN/H2O 9:1 v/v%, flow rate 1.0 mL/min) giving the quinone 4a in pure form (yellowish solid, tR = 4.5 min, 78%). 1H NMR (600 MHz) in CDCl3: δH = 7.29 (2H, d, J = 8.5 Hz), 7.13 (2H, d, J = 8.5 Hz), 6.77 (1H, d, J = 10.2 Hz), 6.71 (1H, dd, J = 10.2, 2.4 Hz), 6.37 (1H, s), 3.71 (2H, s). 13C NMR (175 MHz) in CDCl3: δC = 187.5 (CO), 187.0 (CO), 148.1 (C), 136.7 (CH), 136.4 (CH), 134.8 (C), 133.3 (CH), 133.0 (C), 130.7 (CH), 129.0 (CH), 34.7 (CH2). HRESIMS m/z 233.0359 [M + H]+ (calcd. 233.0364 for C13H10O2Cl).
3.5. Synthesis of Compounds 5a and 5b
Procedure B: The synthesis of compounds
5a and
5b aimed to enlarge the structural diversity of diquinone-derived compounds, following the reported procedure by Love and coworkers [
15]. The general procedure is detailed as follows: CAN (7.0 mmol) was dissolved in H2O (3.50 mL), whereas the arene substrates (1.0 mmol) were dissolved in CH3CN (1.0 mL), and slowly added to the CAN solution. In our case, we performed the oxidation on the 2-(4-bromobenzyl)-1,4-dimethoxybenzene (
3a, 81.8 mg, 0.27 mmol) and the 2-(4-chlorobenzyl)-1,4-dimethoxybenzene (
3b, 88.0 mg, 0.33 mmol). After the addition was complete, the mixture was stirred for 2 h at room temperature, open to the air, observing a precipitate formation. Thus, each mixture was diluted with cold H
2O, and the resulting precipitate was collected by filtration. The resulting orange solid was dried under vacuum and chromatographed by HPLC on Kromasil SiO
2 10 µM column (hexane/EtOAc 8:2
v/
v% and flow rate 3.0 mL/min).
Compound 5a, 4,4′-bis(4-bromobenzyl)-[1,1′-bi(cyclohexane)]-3,3′,6,6′-tetraene-2,2′,5,5′-tetraone: orange solid, tR = 22.9 min (84%). 1H NMR (600 MHz) in CDCl3: δH = 7.46 (2H, d, J = 8.2 Hz), 7.08 (2H, d, J = 8.2 Hz), 6.80 (1H, s), 6.42 (1H, s), 3.72 (2H, s). 13C NMR (175 MHz) in CDCl3: δC = 186.1 (CO), 184.7 (CO), 148.4 (C), 139.3 (C), 136.0 (CH), 134.9 (C), 133.5 (CH), 132.1 (CH), 131.1 (CH), 121.2 (C), 34.6 (CH2). HRESIMS m/z 552.9492 [M + H]+ (calcd. 552.9468 for C26H17O4Br2).
Compound 5b, 4,4′-bis(4-chlorobenzyl)-[1,1′-bi(cyclohexane)]-3,3′,6,6′-tetraene-2,2′,5,5′-tetraone: orange solid, tR = 15.6 min (88%). 1H NMR (600 MHz) in CDCl3: δH = 7.31 (2H, d, J = 8.1 Hz), 7.14 (2H, d, J = 8.1 Hz), 6.80 (1H, s), 6.42 (1H, s), 3.74 (2H, s). 13C NMR (175 MHz) in CDCl3: δC = 186.2 (CO), 184.7 (CO), 148.5 (C), 139.3 (C), 136.0 (CH), 134.4 (C), 133.5 (CH), 133.1 (C), 130.8 (CH), 129.1 (CH), 34.5 (CH2). HRESIMS m/z 463.0482 [M + H]+ (calcd. 463.0498 for C26H17O4Cl2).







{kind=link}