1. Introduction
Carbohydrates are among the most valuable chiral building blocks for synthesizing optically active compounds due to their abundant natural resources. Consequently, numerous examples of natural product syntheses using carbohydrates as chiral building blocks have been documented [
1,
2]. Effective methods to convert carbohydrates into suitable building blocks are essential for these applications. Among naturally occurring sugars, arabinose is unique, with both
d- and
l-arabinose isomers present in nature despite the homochirality of
d-sugars. As both
d- and
l-arabinose are commercially available, arabinose serves as a versatile chiral building block, leading to the synthesis of various derivatives [
3,
4,
5]. Reitz et al. reported a Wittig reaction of
d-arabinose at the anomeric position using diethyl (cyanomethyl)phosphonate and lithium bis(trimethylsilyl)amide, resulting in the E2 elimination of the C3-benzyloxy group to produce a 1,3-diene with a 43% yield [
6]. Additionally, 1-phenylthio-1,3-diene and 1-phenylselenenyl-1,3-diene were obtained in 6% and 55% yields, respectively, using different Wittig reagents [
7,
8]. However, there are no reports on the synthesis of (
S,
Z)-1,4-bis(benzyloxy)hexa-3,5-dien-2-ol (
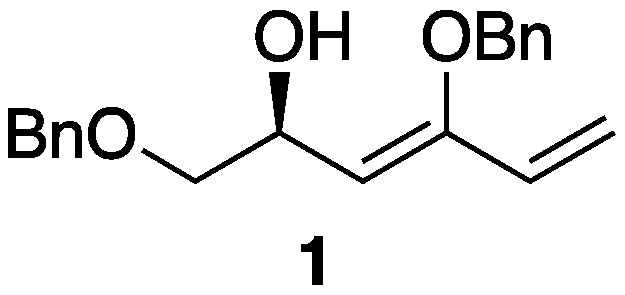
1;
Figure 1), which features a terminal 1,3-diene. The synthesis of 1,3-diene
1 utilized the readily available reagent methyltriphenylphosphonium bromide, resulting in a compound significantly more stable than the previously mentioned 1-substituted 1,3-dienes. Moreover, the terminal 1,3-diene motif is amenable to further transformations, such as cross-metathesis and Diels–Alder reactions. In this paper, we present the synthesis of an optically active 1,3-diene, (
S,
Z)-1,4-bis(benzyloxy)hexa-3,5-dien-2-ol (
1), from 2,3,5-tri-
O-benzyl-
d-arabinofuranose (
3) using methyltriphenylphosphonium bromide and potassium
tert-butoxide in a single step. The cross-metathesis reaction of 1,3-diene
1 with
cis-1,4-diacetoxy-2-butene is also demonstrated.
2. Results and Discussion
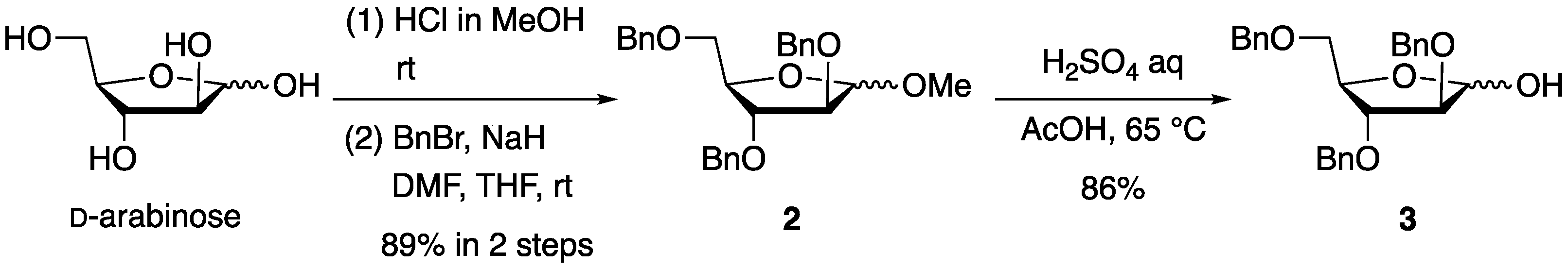
First, 2,3,5-tri-
O-benzyl-
d-arabinofuranose (
3) was prepared from D-arabinose in three steps according to a procedure from the literature with slight modifications (
Scheme 1) [
9].
d-arabinose was treated with methanolic hydrogen chloride to yield methyl
d-arabinofuranoside, and the remaining hydroxy groups were protected with benzyl groups to provide methyl 2,3,5-tri-
O-benzyl-
d-arabinofuranoside (
2) in an 89% yield over two steps. Finally, the treatment of methyl glycoside
2 with aqueous sulfuric acid in acetic acid resulted in the formation of furanose
3 at an 86% yield.
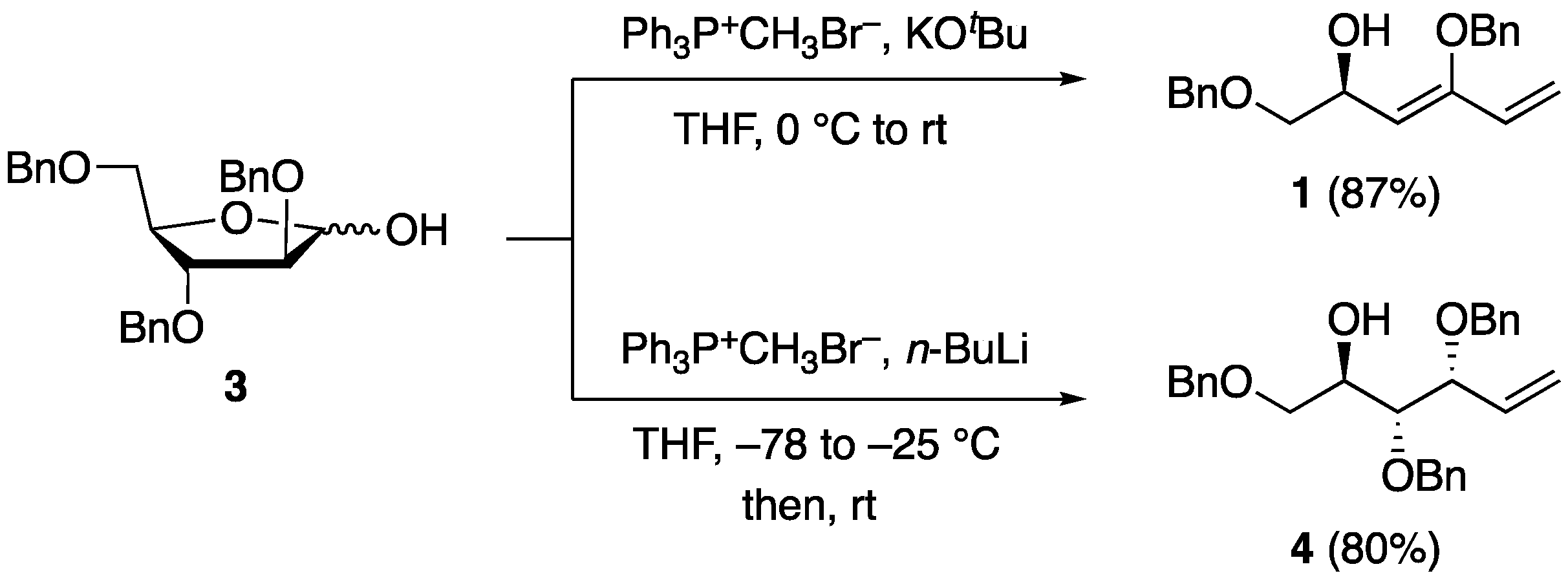
Compound
3 was treated with a ylide prepared from potassium
tert-butoxide and methyltriphenylphosphonium bromide in tetrahydrofuran (THF), yielding 1,3-diene
1 in an 87% yield (
Scheme 2). In this reaction, a Wittig reaction and an elimination reaction of the C3-benzyloxy group occurred to give 1,3-diene
1. A similar elimination/Wittig reaction of 2,3,4,6-tetra-
O-benzyl-
d-glucopyranose has been reported in the literature, although it required a carcinogenic polar solvent, hexamethylphosphoramide [
10]. Interestingly, the reaction of 2,3-
O-isopropylidene-
d-ribofuranose under the same conditions yielded the standard Wittig product without elimination [
11]. Additionally, a Wittig reaction of compound
3 at lower temperatures using methyltriphenylphosphonium bromide and
n-butyl lithium revealed the formation of Wittig product
4 without elimination [
12]. These results indicate that the appropriate conformation of the elimination reaction, the reaction temperature, and the choice of base are crucial for yielding 1,3-diene
1.
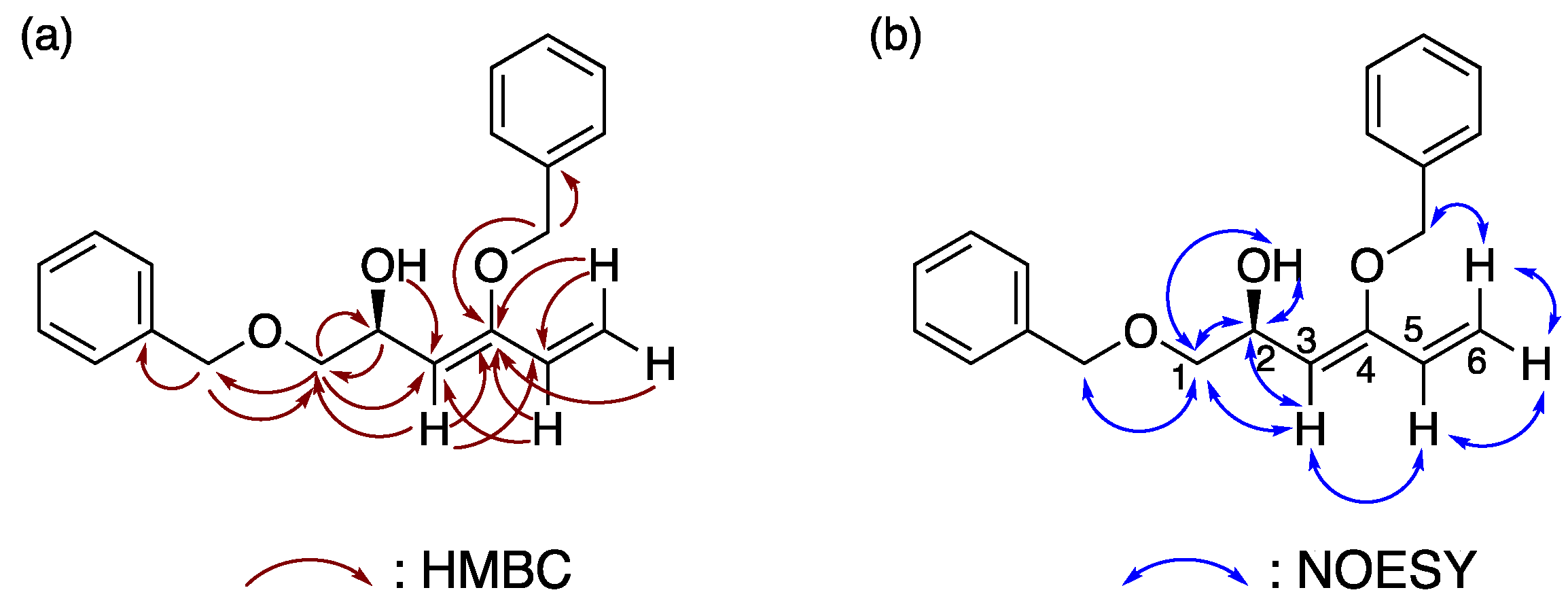
The chemical structure of 1,3-diene
1 was unambiguously determined by HMBC measurements in CDCl
3 in combination with COSY and HSQC spectra (
Figure 2a and
Figures S3–S5). Two key NOESY correlations between H-3 and H-5, and H-6
cis and benzyl protons at the C4 position suggest that the geometry of 1,3-diene was
s-
trans in CDCl
3 solution (
Figure 2b and
Figure S6). The FT-IR spectrum also supported the structure, indicated by the presence of the hydroxy group as a broad band at 3437 cm
−1 and the 1,3-diene moiety as strong bands at 1651 and 1605 cm
−1 (
Figure S7).
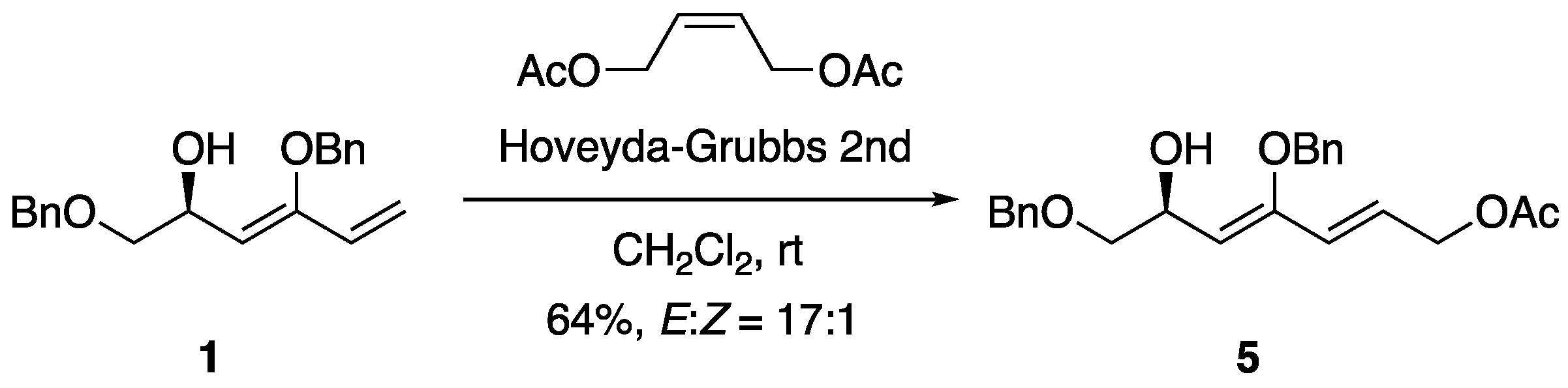
To further investigate the utility of 1,3-diene
1, the cross-metathesis reaction of
1 with
cis-1,4-diacetoxy-2-butene was examined (
Scheme 3). The reaction proceeded smoothly using the Hoveyda–Grubbs second-generation catalyst, yielding metathesis product
5 at a 64% yield with a high
E-selectivity of 17:1 based on
1H NMR analysis. The coupling constants of vicinal olefinic protons of compound
5 were observed at 15.7 Hz for the major
E-isomer and 11.9 Hz for the minor
Z-isomer.
3. Materials and Methods
3.1. General Procedure and Method
Optical rotations were measured on a JASCO DIP-370 polarimeter (JASCO Corporation, Tokyo, Japan) using CHCl
3 as a solvent.
1H NMR and
13C NMR spectra were recorded on a JEOL JNM-ECZ400R (400 MHz and 100 MHz), a Varian Gemini 300 (300 MHz and 75 MHz), or a Varian NMR System 500PS SN (500 MHz and 125 MHz) spectrometers (Agilent Inc., Santa Clara, CA, USA). Chemical shifts (δ) were reported in parts per million (ppm). Tetramethylsilane was used as the internal reference (0.00 ppm in CDCl
3) for
1H NMR spectra, while the central solvent peak was used as a reference (77.0 ppm in CDCl
3) for
13C NMR spectra. The IR spectra were recorded on a Shimadzu IRAffinity-1 FT-IR spectrophotometer (Shimadzu Corporation, Kyoto, Japan). High-resolution mass spectra (HRMS) were obtained on JEOL JMS-T100TD using electrospray ionization (ESI) (JEOL Ltd., Tokyo, Japan) in the time-of-flight (TOF) mode. Analytical thin layer chromatography (TLC) was performed with Merck Millipore precoated TLC plates (MilliporeSigma, Burlington, VT, USA), silica gel at 60 F
254, and layer thicknesses of 0.25 mm. Compounds were observed in UV light at 254 nm and then visualized by staining with iodine,
p-anisaldehyde, or the phosphomolybdic acid stain. Flash and gravity column chromatography separations were performed on Kanto Chemical silica gel 60N, spherical neutral, with particle sizes of 63–210 μm and 40–50 μm, respectively. All moisture-sensitive reactions were conducted under an inert atmosphere. Reagents and solvents were of commercial grade and were used as supplied unless otherwise noted [
13].
3.2. Methyl 2,3,5-Tri-O-benzyl-d-arabinofuranoside (2)
To a solution of acetyl chloride (0.711 mL, 10.0 mmol) in MeOH (100 mL), D-arabinose (3.00 g, 20.0 mmol) was added at room temperature, and the mixture was stirred at room temperature for 18 h. The reaction was quenched by adding Ag2CO3 (2.76 g, 10.0 mmol), and the resultant suspension was stirred at room temperature for 1 h. The precipitate was removed by filtering through a Celite pad, which was washed with MeOH. The combined organics were concentrated under a vacuum. The residue was co-evaporated with toluene to give a crude methyl D-arabinofuranoside (Rf = 0.26 and 0.20, 10% MeOH in CHCl3). To a solution of the above methyl D-arabinofuranoside in THF/DMF (75 mL/25 mL), NaH was added (60% dispersion in mineral oil; 3.60 g, 90.0 mmol) at 0 °C and the reaction mixture was stirred at room temperature for 10 min. Benzyl bromide (10.7 mL, 90.0 mmol) was added to the reaction mixture at 0 °C, which was stirred at room temperature for 19 h. The reaction was quenched by adding water, which was diluted with toluene. After the removal of THF by evaporation, the aqueous layer was extracted with EtOAc three times, and the combined organics were washed with brine and dried over anhydrous MgSO4. The evaporation of the solvent gave a residue, which was purified by flash column chromatography on silica gel (12% EtOAc in n-hexane) to yield the desired product 2 (7.76 g, 89% in 2 steps, α:β = 2.3:1) as a colorless syrup. Rf = 0.63 and 0.59 (40% EtOAc in n-hexane). 1H NMR (300 MHz, CDCl3) δ (α-anomer): 7.43–7.17 (15H, m, ArH), 4.95 (1H, br s, H-1), 4.58–4.51 (4H, m, 4 × CHHPh), 4.48 (1H, d, J = 12.1 Hz, CHHPh), 4.46 (1H,d, J = 11.8 Hz, CHHPh), 4.25–4.17 (1H, m, H-4), 3.99 (1H, dd, J2,3 = 2.8 Hz, J1,2 = 1.1 Hz, H-2), 3.90 (1H,dd, J3,4 = 6.4, J2,3 = 2.8 Hz, H-3), 3.70–3.61 (1H, m, H-5a), 3.61–3.56 (1H, m, H-5b), 3.39 (3H, s, OCH3); δ (β-anomer): 7.43–7.17 (15H, m, ArH), 4.73 (1H, d, J1,2 = 3.6 Hz, H-1), 4.68–4.60 (4H, m, 4 × CHHPh), 4.58–4.51 (2H, m, 2 × CHHPh), 4.15–4.05 (3H, m, H-2, 3, 4), 3.55–3.44 (2H, m, H-5), 3.32 (3H, s, OCH3).
3.3. 2,3,5-Tri-O-benzyl-d-arabinofuranose (3)
To a solution of methyl glycoside 2 (7.76 g, 17.9 mmol) in AcOH (180 mL), 2 M H2SO4 aq was added (90 mL) at 65 °C, and the reaction mixture was stirred at the same temperature for 41 h. The mixture was diluted with water and extracted with CH2Cl2 three times. The combined organics were successively washed with water, sat. NaHCO3 aq, and water. After drying with MgSO4, the residue was purified by flash column chromatography on silica gel (20% EtOAc in n-hexane) to yield the desired product 3 (6.44 g, 86%, α:β = 1.2:1) as white solids along with the recovery of 2 (445 mg, 6% rsm). Rf = 0.41 (40% EtOAc in n-hexane). 1H NMR (300 MHz, CDCl3) δ (α-anomer): 7.41–7.20 (15H, m, ArH), 5.39 (1H, s, H-1), 4.69–4.41 (7H, m, H-4, 6 × CHHPh), 3.99–3.91 (2H, m, H-2, 3), 3.59 (1H, dd, J5a,5b = 10.2, J4,5a = 6.6 Hz, H-5a), 3.56–3.46 (1H, m, H-5b); δ (β-anomer): 7.41–7.20 (15H, m, ArH), 5.33 (1H, d, J1,2 = 4.3 Hz, H-1), 4.69–4.41 (6H, m, 6 × CHHPh), 4.16 (1H, t, J2,3 = J3,4 = 4.7 Hz, H-3), 4.12–4.05 (1H, m, H-4), 4.01 (1H, dd, J2,3 = 4.7 Hz, J1,2 = 4.3 Hz, H-2), 3.56–3.46 (2H, m, H-5).
3.4. (S,Z)-1,4-Bis(benzyloxy)hexa-3,5-dien-2-ol (1)
To a suspension of methyltriphenylphosphonium bromide (598 mg, 1.66 mmol) in THF (8 mL), potassium tert-butoxide was added (186 mg, 1.66 mmol) at 0 °C, and the resultant mixture was stirred for 1 h at 0 °C and for 1 h at room temperature. Then, a solution of 2,3,5-tri-O-benzyl-D-arabinofuranose (3; 200 mg, 0.476 mmol) in THF (2 mL) was dropped into the reaction mixture at 0 °C and the reaction mixture was stirred at room temperature for 1.5 h. The reaction was quenched by adding water, and the aqueous phase was extracted with EtOAc (3 times). The combined organics were washed with brine, dried over anhydrous Na2SO4, and concentrated in vacuo. The residue was purified by flash column chromatography on silica gel (10% EtOAc in n-hexane) to yield 1,3-diene 1 (129 mg, 87%) as a colorless oil. Rf = 0.35 (30% EtOAc in n-hexane). [α]28D + 2.22 (c 1.00, CHCl3). 1H NMR (400 MHz, CDCl3) δ: 7.51–7.28 (10H, m, ArH), 6.14 (1H, dd, J = 17.4, 10.8 Hz, H-5), 5.52 (1H, dd, J = 17.4, 1.4 Hz, H-6cis), 5.21 (1H, dd, J = 10.8, 1.4 Hz, H-6trans), 5.08 (1H, d, J = 8.3 Hz, H-3), 4.84 (1H, d, J = 11.5 Hz, ArCHHOC=C), 4.80 (1H, d, J = 11.5 Hz, ArCHHOC=C), 4.71 (1H, dddd, J = 8.3, 7.6, 3.6, 3.1 Hz, H-2), 4.51 (2H, s, ArCH2OCH2), 3.38 (1H, dd, J = 9.7, 3.6 Hz, H-1a), 3.31 (1H, dd, J = 9.7, 7.6 Hz, H-1b), 2.36 (1H, d, J = 3.1 Hz, OH). 13C{1H} NMR (100 MHz, CDCl3) δ: 154.9 (C-4), 137.9 (C-Ar next to -CH2OCH2-), 137.2 (C-Ar next to -CH2OC=C-), 131.9 (C-5), 128.5 (2C, C-Ar), 128.4 (2C, C-Ar), 128.10 (C-Ar), 128.08 (2C, C-Ar), 127.7 (3C, C-Ar), 117.0 (C-3), 116.1 (C-6), 73.6 (ArCH2OC=C-), 73.5 (C-1), 73.2 (ArCH2OCH2-), 65.9 (C-2). IR (KBr): 3537 (br), 3090, 3063, 3032, 2916, 2860, 1651, 1605 cm−1. HRMS (ESI) m/z: [M + Na]+ calcd for C20H22O3Na, 333.1467; found, 333.1466.
3.5. Cross-Metathesis Product 5
To a solution of 1,3-diene 1 (62.1 mg, 0.200 mmol) and cis-1,4-diacetoxy-2-butene (158 mg, 1.00 mmol) in CH2Cl2 (2.0 mL), a Hoveyda–Grubbs second-generation catalyst (25.1 mg, 0.0400 mmol) was added at room temperature under argon atmosphere, and the mixture was stirred at room temperature for 60 min. The reaction mixture was filtered through a short pad of amino silica gel (up)/flash silica gel (bottom) with CHCl3 and the solvent was removed under reduced pressure. The residue was purified by flash column chromatography on silica gel (30% EtOAc in n-hexane) to give the desired product 5 (49.0 mg, 64%, E/Z = 17:1) as a colorless oil. Rf = 0.19 (30% EtOAc in n-hexane). 1H NMR (500 MHz, CDCl3) δ: 7.38–7.29 (10H, m), 6.11–5.98 (2H, m), 5.11 (1H, d, J = 8.6 Hz), 4.81 (2H, d, J = 2.9 Hz), 4.73–4.68 (1H, m), 4.64 (2H, d, J = 5.1 Hz), 4.51 (2H, s), 3.38 (1H, dd, J = 9.7, 3.5 Hz), 3.31 (1H, dd, J = 9.7, 7.6 Hz), 2.39 (1H, br s), 2.08 (3H, s). 13C{1H} NMR (125 MHz, CDCl3) δ: 170.6, 153.8, 137.8, 137.0, 128.5 (3C), 128.4 (2C), 128.2, 128.0 (2C), 127.7 (3C), 125.4, 117.8, 73.9, 73.4, 73.2, 65.8, 64.0, 20.9. HRMS (ESI) m/z: [M + Na]+ calcd for C23H26O5Na, 405.1678; found, 405.1669.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}