
4-(Tris(4-methyl-1H-pyrazol-1-yl)methyl)aniline


 , , , and
, , , and
Abstract

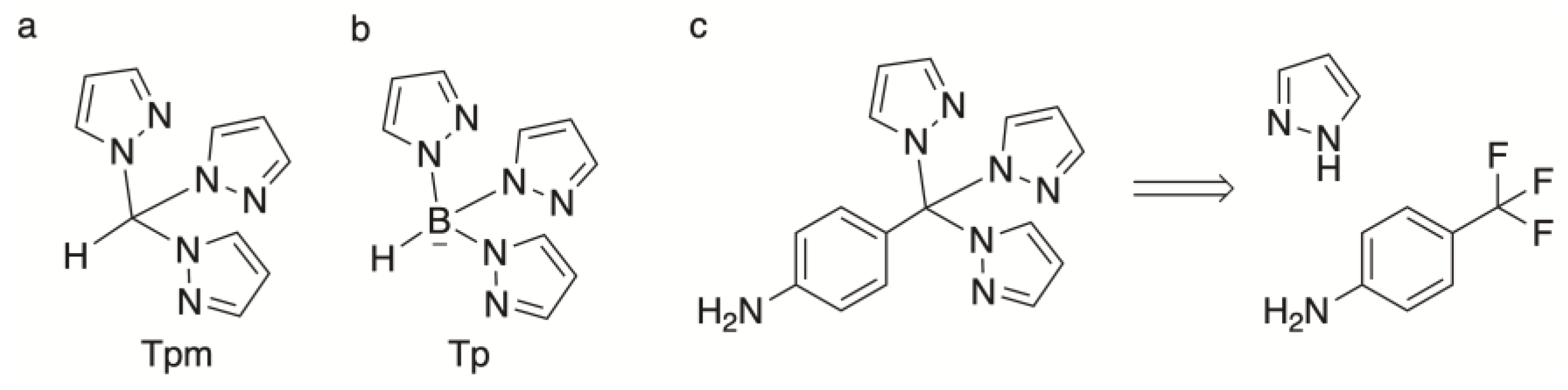
1. Introduction
2. Results and Discussion
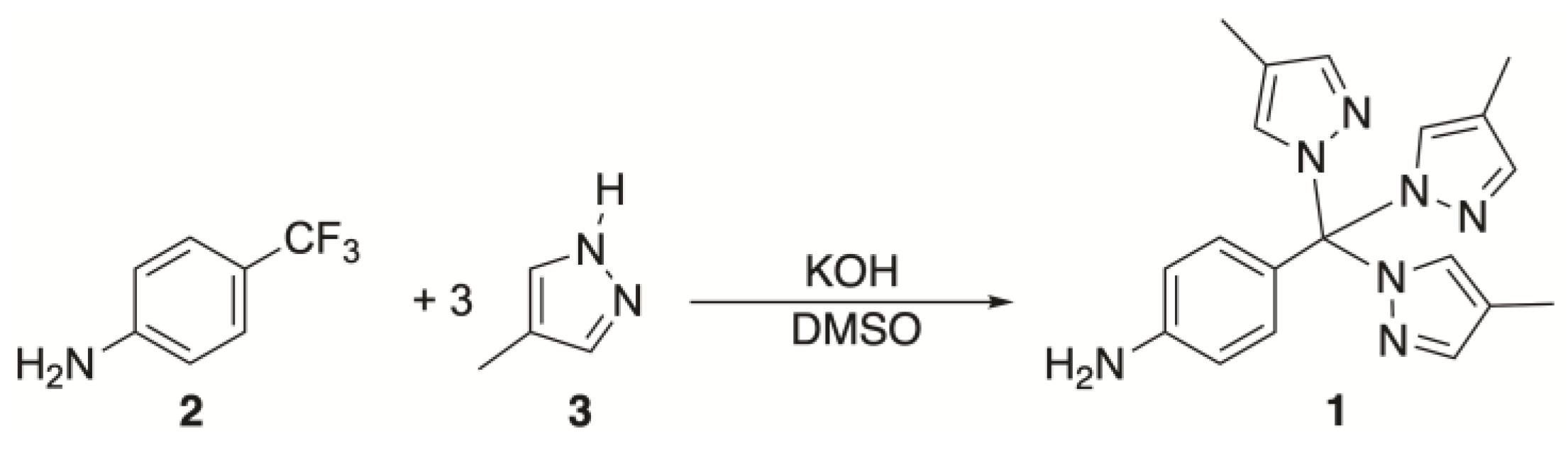
2.1. Synthesis
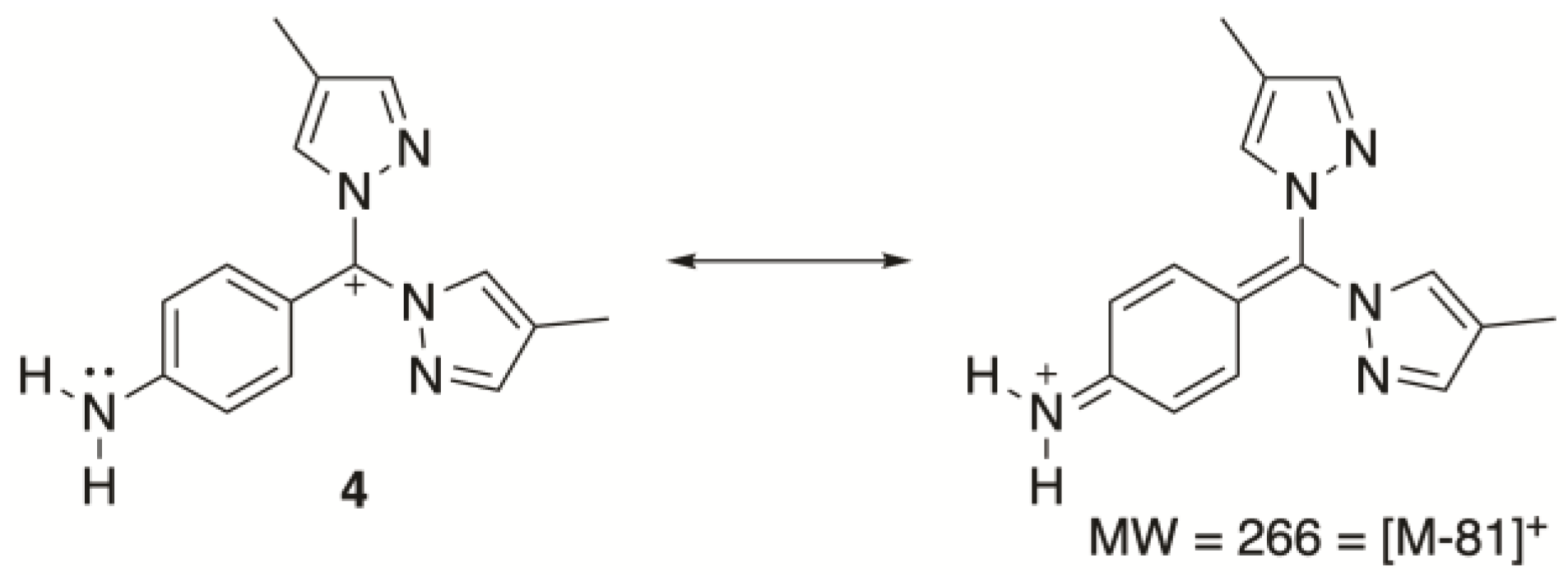
2.2. Characterization
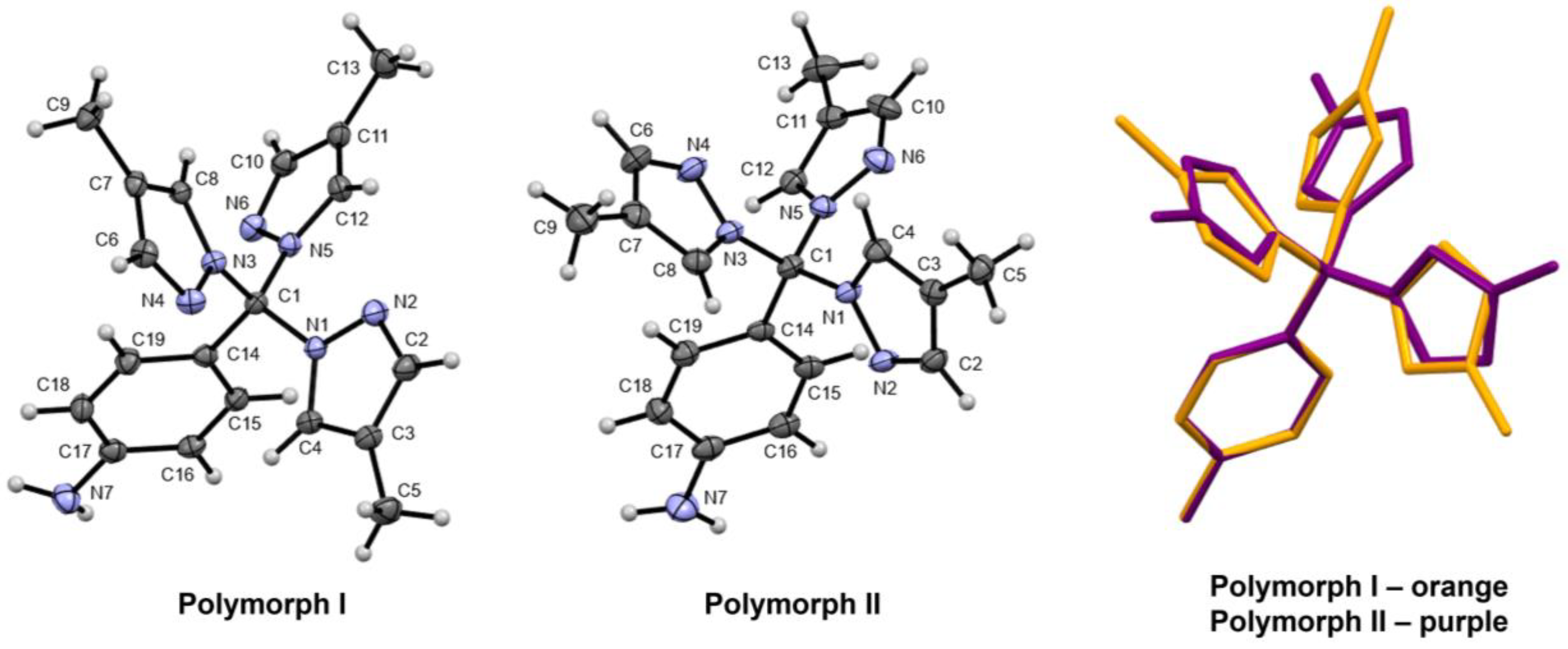
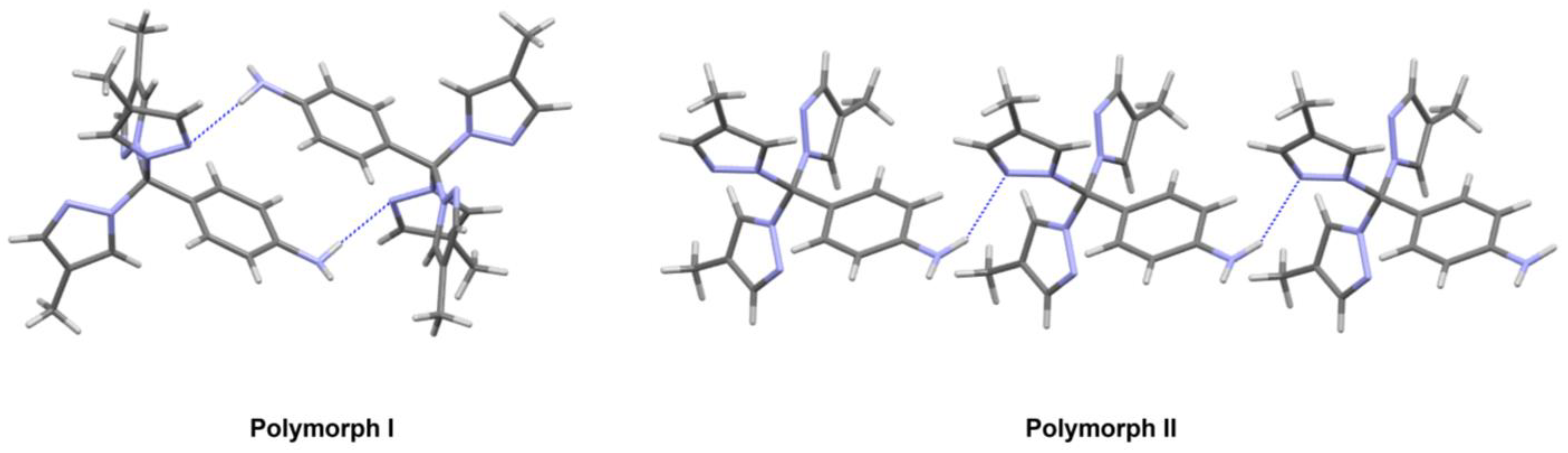
2.3. Solid-State Structure and Polymorphism
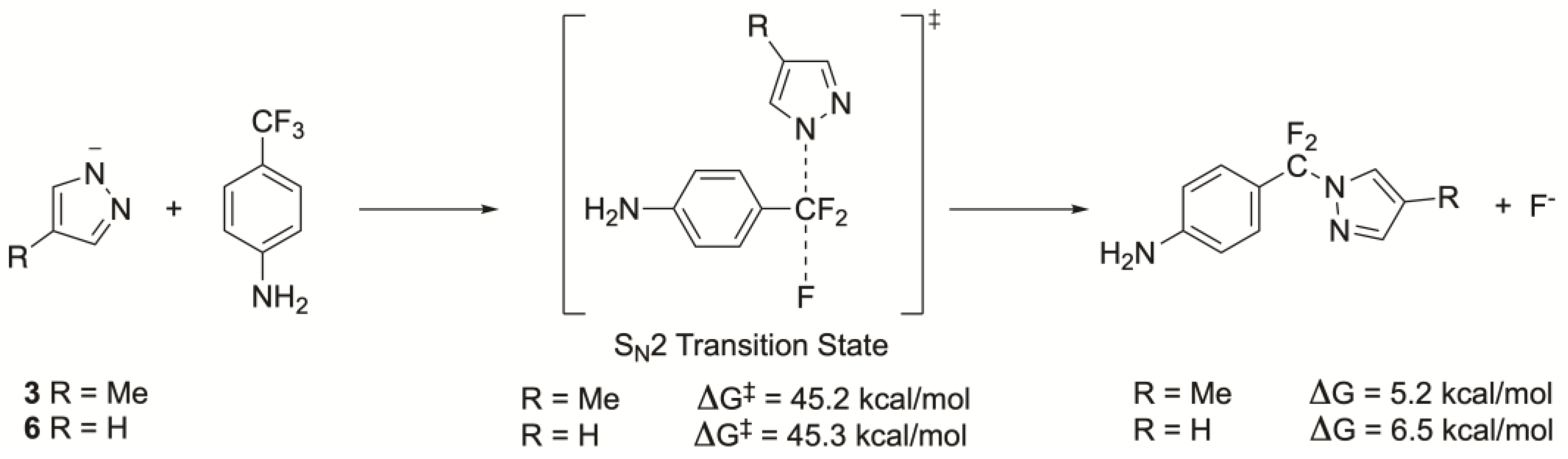
2.4. Computational Studies
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Martins, L.M.D.R.S. C-scorpionate complexes: Ever young catalytic tools. Coord. Chem. Rev. 2019, 396, 89–102. [Google Scholar] [CrossRef]
- Martins, L.M.D.R.S.; Pombeiro, A.J.L. Water-Soluble C-Scorpionate Complexes—Catalytic and Biological Applications. Eur. J. Inorg. Chem. 2016, 2016, 2236–2252. [Google Scholar] [CrossRef]
- Muñoz-Molina, J.M.; Belderrain, T.R.; Pérez, P.J. Group 11 tris(pyrazolyl)methane complexes: Structural features and catalytic applications. Dalton Trans. 2019, 48, 10772–10781. [Google Scholar] [CrossRef] [PubMed]
- Niesel, J.; Pinto, A.; N’Dongo, H.W.P.; Merz, K.; Ott, I.; Gust, R.; Schatzschneider, U. Photoinduced CO release, cellular uptake and cytotoxicity of a tris(pyrazolyl)methane (tpm) manganese tricarbonyl complex. Chem. Commun. 2008, 1798–1800. [Google Scholar] [CrossRef]
- Trofimenko, S. Boron-Pyrazole Chemistry. J. Am. Chem. Soc. 1966, 88, 1842–1844. [Google Scholar] [CrossRef]
- Caballero, A.; Díaz-Requejo, M.M.; Fructos, M.R.; Urbano, J.; Pérez, P.J. Modern Applications of Trispyrazolylborate Ligands in Coinage Metal Catalysis. In Ligand Design in Metal Chemistry; Wiley: Hoboken, NJ, USA, 2016; pp. 308–329. [Google Scholar]
- Theopold, K.H.; Reinaud, O.M.; Doren, D.; Konecny, R. Dioxygen activation with sterically hindered tris(pyrazolyl)borate cobalt complexes. In Studies in Surface Science and Catalysis; Grasselli, R.K., Oyama, S.T., Gaffney, A.M., Lyons, J.E., Eds.; Elsevier: Amsterdam, The Netherlands, 1997; Volume 110, pp. 1081–1088. [Google Scholar]
- Slugovc, C.; Padilla-Martínez, I.; Sirol, S.; Carmona, E. Rhodium- and iridium-trispyrazolylborate complexes: C-H activation and coordination chemistry. Coord. Chem. Rev. 2001, 213, 129–157. [Google Scholar] [CrossRef]
- Bromberg, S.E.; Yang, H.; Asplund, M.C.; Lian, T.; McNamara, B.K.; Kotz, K.T.; Yeston, J.S.; Wilkens, M.; Frei, H.; Bergman, R.G.; et al. The Mechanism of a C-H Bond Activation Reaction in Room-Temperature Alkane Solution. Science 1997, 278, 260–263. [Google Scholar] [CrossRef]
- Trofimenko, S. Geminal poly(1-pyrazolyl)alkanes and their coordination chemistry. J. Am. Chem. Soc. 1970, 92, 5118–5126. [Google Scholar] [CrossRef]
- Juliá, S.; del Mazo, J.M.; Avila, L.; Elguero, J. Improved Synthesis of Polyazolylmethanes Under Solid-Liquid Phase-Transfer Catalysis. Org. Prep. Proced. Int. 1984, 16, 299–307. [Google Scholar] [CrossRef]
- Schorpp, M.; Heizmann, T.; Schmucker, M.; Rein, S.; Weber, S.; Krossing, I. Synthesis and Application of a Perfluorinated Ammoniumyl Radical Cation as a Very Strong Deelectronator. Angew. Chem. Int. Ed. 2020, 59, 9453–9459. [Google Scholar] [CrossRef]
- Liddle, B.J.; Gardinier, J.R. A Practical Synthesis of Tris(pyrazolyl)methylaryls. J. Org. Chem. 2007, 72, 9794–9797. [Google Scholar] [CrossRef] [PubMed]
- McDarmont, S.L.; McMillen, C.D.; Temelso, B.; Pienkos, J.A. Exploiting a C–F Activation Strategy to Generate Novel Tris(pyrazolyl)methane Ligands. Z. Für Anorg. Und Allg. Chem. 2020, 646, 1886–1891. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef] [PubMed]
- Bordwell, F.G.; Algrim, D.J. Acidities of anilines in dimethyl sulfoxide solution. J. Am. Chem. Soc. 1988, 110, 2964–2968. [Google Scholar] [CrossRef]
- Bordwell, F.G. Equilibrium acidities in dimethyl sulfoxide solution. Acc. Chem. Res. 1988, 21, 456–463. [Google Scholar] [CrossRef]
- Gottlieb, H.E.; Kotlyar, V.; Nudelman, A. NMR Chemical Shifts of Common Laboratory Solvents as Trace Impurities. J. Org. Chem. 1997, 62, 7512–7515. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Rev. C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Yu, H.S.; He, X.; Li, S.L.; Truhlar, D.G. MN15: A Kohn–Sham global-hybrid exchange–correlation density functional with broad accuracy for multi-reference and single-reference systems and noncovalent interactions. Chem. Sci. 2016, 7, 5032–5051. [Google Scholar] [CrossRef] [PubMed]
- Daga, L.E.; Civalleri, B.; Maschio, L. Gaussian Basis Sets for Crystalline Solids: All-Purpose Basis Set Libraries vs System-Specific Optimizations. J. Chem. Theory Comput. 2020, 16, 2192–2201. [Google Scholar] [CrossRef]
- Yu, H.S.; He, X.; Truhlar, D.G. MN15-L: A New Local Exchange-Correlation Functional for Kohn–Sham Density Functional Theory with Broad Accuracy for Atoms, Molecules, and Solids. J. Chem. Theory Comput. 2016, 12, 1280–1293. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2017; Available online: https://www.R-project.org/ (accessed on 27 March 2024).
- Bruker. APEX4; Bruker AXS Inc.: Madison, WI, USA, 2017. [Google Scholar]
- Sheldrick, G. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Macrae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M.; et al. Mercury 4.0: From visualization to analysis, design and prediction. J. Appl. Crystallogr. 2020, 53, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Paton, A.S.; Lough, A.J.; Bender, T.P. One Well-Placed Methyl Group Increases the Solubility of Phenoxy Boronsubphthalocyanine Two Orders of Magnitude. Ind. Eng. Chem. Res. 2012, 51, 6290–6296. [Google Scholar] [CrossRef]
- Pinheiro, P.S.M.; Franco, L.S.; Fraga, C.A.M. The Magic Methyl and Its Tricks in Drug Discovery and Development. Pharmaceuticals 2023, 16, 1157. [Google Scholar] [CrossRef]
- Reger, D.L. Tris(Pyrazolyl)Methane Ligands: The Neutral Analogs of Tris(Pyrazolyl)Borate Ligands. Comments Inorg. Chem. 1999, 21, 1–28. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Rings | Polymorph I | Polymorph II |
|---|---|---|
| A–B | 87.96 (5) | 73.20 (7) |
| A–C | 22.66 (10) | 57.19 (11) |
| B–C | 83.37 (4) | 79.06 (10) |
| A–D | 78.16 (5) | 56.80 (10) |
| B–D | 80.60 (5) | 81.78 (10) |
| C–D | 79.78 (5) | 79.77 (10) |
| Polymorph | D-H (Å) | H···A (Å) | D···A (Å) | D-H···A (°) |
|---|---|---|---|---|
| I | ||||
| N7-H7N2···N6 a | 0.92 (2) | 2.33 (2) | 3.2463 (19) | 178.2 (18) |
| C10-H10···N4 b | 0.95 | 2.65 | 3.5450 (19) | 157.0 |
| II | ||||
| N7-H7N2···N6 c | 0.94 (3) | 2.33 (3) | 3.199 (3) | 152 (2) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garrison, B.B.; Duhamel, J.E.; Antoine, N.; Symes, S.J.K.; Grice, K.A.; McMillen, C.D.; Pienkos, J.A. 4-(Tris(4-methyl-1H-pyrazol-1-yl)methyl)aniline. Molbank 2024, 2024, M1823. https://doi.org/10.3390/M1823
Garrison BB, Duhamel JE, Antoine N, Symes SJK, Grice KA, McMillen CD, Pienkos JA. 4-(Tris(4-methyl-1H-pyrazol-1-yl)methyl)aniline. Molbank. 2024; 2024(2):M1823. https://doi.org/10.3390/M1823
Chicago/Turabian StyleGarrison, Bradley B., Joseph E. Duhamel, Nehemiah Antoine, Steven J. K. Symes, Kyle A. Grice, Colin D. McMillen, and Jared A. Pienkos. 2024. "4-(Tris(4-methyl-1H-pyrazol-1-yl)methyl)aniline" Molbank 2024, no. 2: M1823. https://doi.org/10.3390/M1823
APA StyleGarrison, B. B., Duhamel, J. E., Antoine, N., Symes, S. J. K., Grice, K. A., McMillen, C. D., & Pienkos, J. A. (2024). 4-(Tris(4-methyl-1H-pyrazol-1-yl)methyl)aniline. Molbank, 2024(2), M1823. https://doi.org/10.3390/M1823