1. Introduction
We have developed an efficient synthetic protocol for the selective preparation of 3,5-disubstituted isoxazoles starting from activated primary nitro compounds and alkynes. The reaction is usually conducted in water or chloroform and involves the use of a simple and inexpensive catalyst, such as sodium hydroxide or 1,4-diazabicyclo[2.2.2]octane (DABCO) [Equation (1)] [
1,
2].
Before this innovation, nitro compounds and alkynes involved in this process were combined with a dehydrating agent, the most popular of which was phenyl isocyanate [
3]. The dehydrating agent was used in a stoichiometric amount and the process was intrinsically incompatible with water.
Our catalyzed reaction has been studied with a wide range of functionalized and non-functionalized terminal alkynes [
4]. Extension of the protocol to the synthesis of 5-methylisoxazoles and the corresponding open derivatives, 2-acetyl enamines, would involve the use of volatile propyne (b.p. = −23 °C) [R = Me, Equation (1)]. In this case, the thermal reaction, which can be performed in a sealed tube, would present safety and scalability difficulties. Thus, for practical reasons, we planned to use a potential synthetic equivalent of propyne, such as propagylbenzoate [R = CH
2OC(O)Ph, Equation (1)]).
The retrosynthetic approach is based on catalytic hydrogenation to convert the primary adduct, isoxazol-5-ylmethyl benzoate, to the desired 5-methylisoxazole (
Scheme 1). This protocol presents two challenges: (i) the use of the methyleneisoxazole group [i.e., an isoxazole ring attached to a methylene (–CH
2) moiety] as a benzyl-like group; and (ii) performing selective hydrogenolysis of the benzyl-like group in the presence of a reducible isoxazole N–O bond.
Numerous examples of catalytic hydrogenation of isoxazole derivatives are reported in the literature. Among them, in most cases the isoxazole ring is not affected by hydrogenolysis under mild conditions using palladium as a metal catalyst (for representative examples of debenzylation, nitro group reduction, and double bond hydrogenation see page 2297 of [
5], page 5/6 of [
6], and page S14 of [
7]), and only in some cases the ring opens to give an enaminone [
8].
Concerning the benzyl-like properties of the methyleneisoxazole group with respect to hydrogenolysis, only one example is described in the literature. In particular, a 5-(acetamidomethyl)isoxazole is converted into a 2:1 mixture of denitrogenated enaminone and 5-methyl-isoxazole derivatives by treatment with H
2 in the presence of 5% Pd/C [
9] (p. 770).
We describe here the hydrogenation over Pd/C of ethyl 5-(benzoyloxymethyl)isoxazole-3-carboxylate. The benzyl-like behavior of the methyleneisoxazole group was confirmed because only a deoxygenated product was observed. Notably, with this substrate it was not possible to isolate the intermediate 5-methylisoxazole since a domino process of two sequential reductions occurred.
2. Results and Discussion
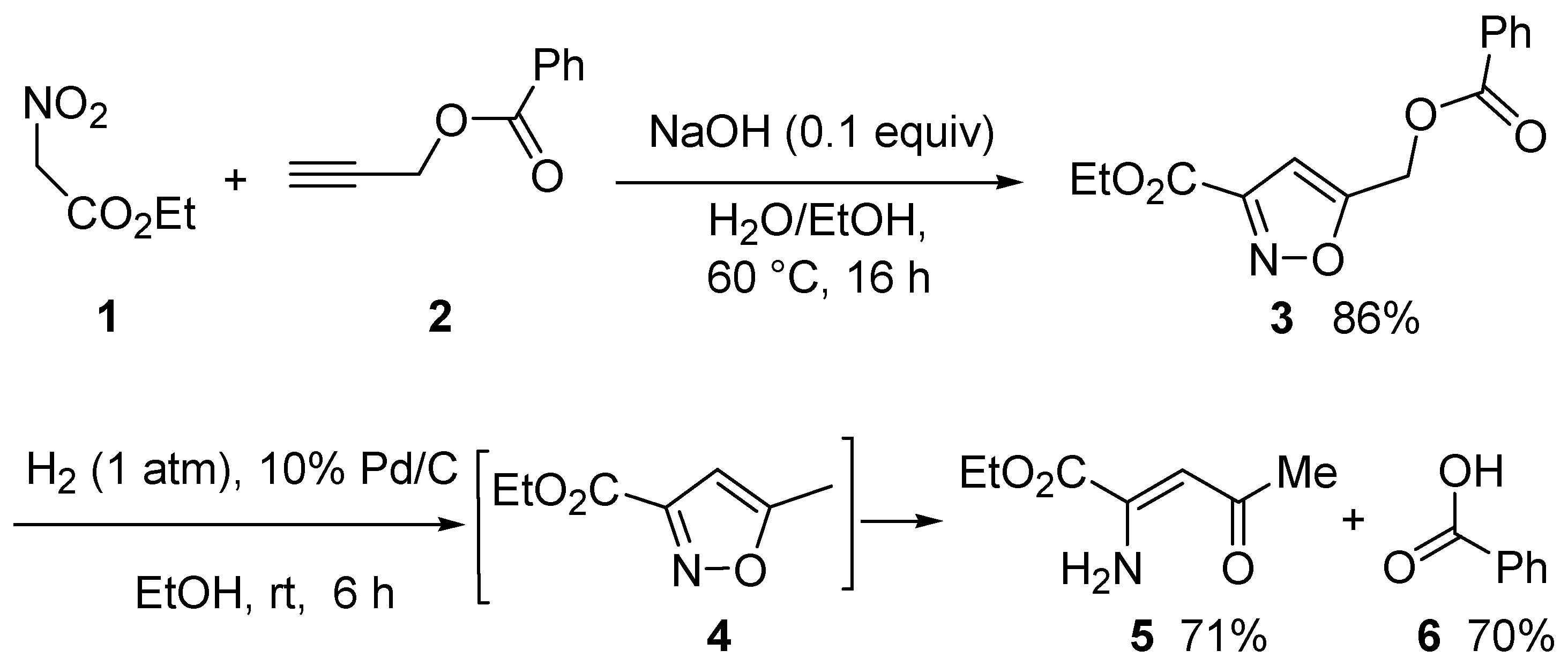
To verify the above idea, we first prepared the undescribed isoxazole (
3) via cycloaddition-condensation of propargyl benzoate (
2) with ethyl nitroacetate (
1). The reaction was conducted in water at a temperature of 60 °C using sodium hydroxide as a catalyst. The cycloadduct (
3) was easily purified by standard silica gel chromatography and was obtained in high yield (
Scheme 2).
With isoxazole (
3) in hand, a series of palladium-catalyzed hydrogenations were carried out using different amounts of Pd (from 10 to 30 mol%) and various solvents with different polarities (ethyl acetate, tetrahydrofuran, and ethanol) (
Table S1). The reactions were conducted at room temperature over a pressure of one atmosphere of hydrogen. Enaminone (
5) was the only product obtained in all cases, with varying degrees of conversion of
3.
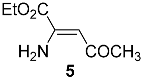
The 1H and 13C NMR spectra in CDCl3 of 5 showed the presence of only one isomer.
The configurations of
E- and
Z-enaminones are commonly assigned considering that a strong hydrogen bond between N–H and the carbonyl occurs in
Z diastereomers, causing the
1H–N shift to move to a higher frequency [
10]. For
E-isomers, an analogous intramolecular hydrogen bond is not possible. Accordingly, the presence of two distinct
1H–N broad singlets at 5.77 and 8.97 ppm in the
1H NMR spectrum proved that
5 was formed as a Z isomer with complete diastereoselectivity. As expected, the two N–H protons in the
E isomer were isochronous and resonated at 6.25 ppm as a single broad singlet [
11]. Interestingly, the
13C NMR spectra of
5 and its
E isomer were also quite different (
Table 1). In particular, the resonance of the methyl carbon in
5 was shifted downfield by 7.5 ppm (30.3 vs. 22.8 ppm of
5 compared to
E-enaminone), and the alkene carbons resonated at 96.8 and 145.5 ppm for
Z-enaminone
5 and at 94.2 and >160 ppm for
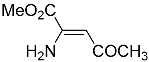
E-enaminone. Furthermore, Mitani et al. [
11] reported that they isolated
E-enaminone as an oil, while
5 was a solid with a melting point of 38.5–39.5 °C. On the contrary, the
1H and
13C NMR spectra of
5 and the corresponding
Z methyl ester reported by Terent’ev et al. [
12] were identical except for the carbinol signals (
Table 1).
The experimental data confirmed that the methyleneisoxazole group indeed acted as a benzyl-like group because the deoxygenation by hydrogenolysis was complete. Under the applied conditions, 5-methylisoxazole (4) spontaneously undergoes N–O bond opening to enaminone (5). Since the formation of the benzoyloxy enaminone was never observed, we can conclude that hydrogenolysis of the methyleneisoxazole group was faster than opening of the isoxazole ring.
3. Materials and Methods
Melting points were determined in open capillary tubes using a Stuart Scientific SMP3 melting point apparatus. Chromatographic separations were performed on silica gel 60 (40–6.3 mm) with analytical grade solvents, driven by a positive pressure of air; R
f values refer to TLC (visualized with UV light and/or by dipping the plates into an acidic solution of vanillin followed by heating with a heat gun). TLC was carried out on alumina-backed plates coated with 25 mm silica gel (Merck F254) with the same eluant as indicated for the column chromatography. The notation PE refers to petroleum ether fraction boiling between 40 and 60 °C. Solvent removal was performed by evaporation under reduced pressure at room temperature.
1H NMR spectra were recorded on a Varian Mercury Plus 400 spectrometer operating at 400 MHz.
13C NMR spectra were recorded on a Varian Gemini 200 spectrometer operating at 50.3 MHz. The
1H NMR data are reported as s = singlet, d = doublet, t = triplet, m = multiplet or unresolved, and br = broad signal, coupling constant(s) in Hz, integration. Multiplicity of the
13C NMR signals and assignments were determined by means of gHMQC and gHMBC experiments. Chemical shifts were determined relative to the residual solvent peak (CHCl
3: 7.24 ppm for
1H NMR and 77.0 ppm for
13C NMR); ESI (electrospray ionization) mass spectra were recorded (infusing the sample solution directly into the ESI chamber by syringe pump) on a Thermo Fisher LCQ-Fleet ion-trap instrument, and spectra were recorded using the ESI
+ technique. Ion mass/charge ratios (
m/
z) are reported as values in atomic mass units followed by the intensities relative to the base peak in parentheses. IR spectra were recorded on an IRAaffinity-1S Shimadzu spectrophotometer; bands are characterized as broad (br), strong (s), medium (m), and weak(w). Elemental analyses were performed with a Thermo Scientific FlashSmart CHNS/O Elemental Analyzer. Propargyl benzoate (
2) was prepared from propargyl alcohol with benzoylchloride in the presence of triethyl amine following a described procedure [
13]. Palladium on charcoal was purchased from Merck (Darmstadt, Germany).
Synthesis of ethyl 5-(benzoyloxymethyl)isoxazole-3-carboxylate (3): A solution of NaOH (4.24 M, 0.040 mL, 0.170 mmol) was added to a mixture of propargyl benzoate (
2) (272 mg, 1.70 mmol), ethyl nitroacetate (
1) (564 mg, 4.24 mmol), water (4160 mg), and ethanol (1280 mg), and the mixture was vigorously stirred in a sealed tube at 60 °C for 16 h. The reaction mixture was concentrated and the residue was subjected to flash chromatography on silica gel (eluant PE/AcOEt = 5:1 containing 3% of Et
3N) to give isoxazole
3 (R
f = 0.36) as clear oil (402 mg, 86% yield).
1H NMR (CDCl
3, Figure S1): δ = 1.39 (t,
J =7.2 Hz, 3 H, C
H3CH
2), 4.43 (q, 2 H;
J =7.2 Hz; CH
3C
H2), 5.45 (s; 2 H, C
H2O), 6.78 (s, 1 H; 4-H), 7.42 (tm, 2 H,
J =7.8 Hz, Ph-H), 7.58 (t, 1 H,
J = 7.8 Hz, Ph-H), 8.02 ppm (dm, 2 H,
J = 7.8 Hz, Ph-H);
13C NMR (CDCl
3,
Figure S1): δ = 14.0 (q, CH
2CH
3), 56.5 (t,
CH
2), 62.3 (t, CH
3CH
2), 104.7 (d, C-4), 128.5 (d, 2 C, Ph-C), 128.8 (s, Ph-C), 129.8 (d, 2 C, Ph-C); 133.6 (d, Ph-C), 156.5 (s, C-3), 159.5 (s,
CO), 165.6 (s,
CO), 168.6 ppm (s, C-5); IR (KBr): ν 3142 (w), 2983 (w), 1728 (s) [C=O], 1600 (m) [C=N], 1471 (m), 1452 (m), 1249 (s), 1201 (s), 1095 (s), 1070 (s), 1026 (s), 779 (m), 711 (s) cm
−1; MS (ESI
+, MeOH):
m/z (%) = 276 (100) [M+1]
+; Elemental analysis calcd (%) for C
14H
13NO
5 (275.26): C 61.09, H 4.76, N 5.09; found: C 60.91, H 4.61 N 4.82.
Synthesis of ethyl (Z)-2-amino-4-oxo-2-pentanoate (5): Pd/C (10%, 79 mg), 3 (102 mg, 0.37 mmol), and anhydrous EtOH (8 mL) were placed in a flask. Then, the reaction mixture was placed under an atmosphere of hydrogen using a balloon.
The suspension was stirred at ambient temperature until starting enaminone (3) disappeared on a TLC control (6 h, eluant: PE/AcOEt = 2:1, Rf = 0.67). After filtration and evaporation of EtOH, the residue was subjected to column chromatography on silica gel (eluant: PE/AcOEt = 7:1 containing 3% of Et3N, then MeOH) to afford 5 (41 mg, Rf = 0.42, 71% yield) as white solid and then benzoic acid triethylammonium salt (6·Et3N, 59 mg, 70%).
5: m.p. 38.5–39.5 °C (Lit. [
14] m. p. 39 °C);
1H NMR (CDCl
3,
Figure S2): δ = 1.34 (t,
J = 7.2 Hz, 3 H, C
H3CH
2), 2.16 (s, 3 H, C
H3CO), 4.30 (q,
J = 7.2 Hz, 2 H, CH
3C
H2), 5.77 (br s, 1 H, N
H), 5.89 (s, 1 H, C=C
H), 8.97 ppm (br s, 1 H, N
H);
13C NMR (CDCl
3,
Figure S2): δ 14.2 (q, CH
2CH
3), 30.3 (q, CO
CH
3), 62.5 (t, CH
3CH
2), 96.8 (d, C=
CH), 145.5 (s,
C=CH), 163.9 (s,
CO
2Et), 199.6 ppm (s,
COCH
3); IR (KBr): ν 3431 (s) [N–H], 3287 (m) [N–H], 3172 (w) [=C–H], 3115 (w) [=C–H], 2997 (w) [C–H], 2974 (w) [C–H], 2941 (w) [C–H], 1716 (s) [C=O], 1645 (m) [C=O], 1589 (m), 1539 (m), 1361 (m), 1278 (s), 1024 (m), 768 (m) cm
−1; MS (ESI
+, MeOH):
m/z (%) = 180 (100) [M + Na]
+; Elemental analysis calcd (%) for C
7H
11NO
3 (157.169): C 53.49, H 7.05, N 8.91; found: C 53.64, H 6.99 N 8.64.
6·Et3N: 1H NMR (CDCl3): δ = 1.30 (t, J = 7.4 Hz, 9 H, 3 × CH3CH2), 3.10 (q, J = 7.4 Hz, 6 H, 3 × CH3CH2), 7.38 (m, 3 H, Ph-H), 8.04 ppm (dd, J = 8.2 and 1.2 Hz, 2 H, Ph-H).





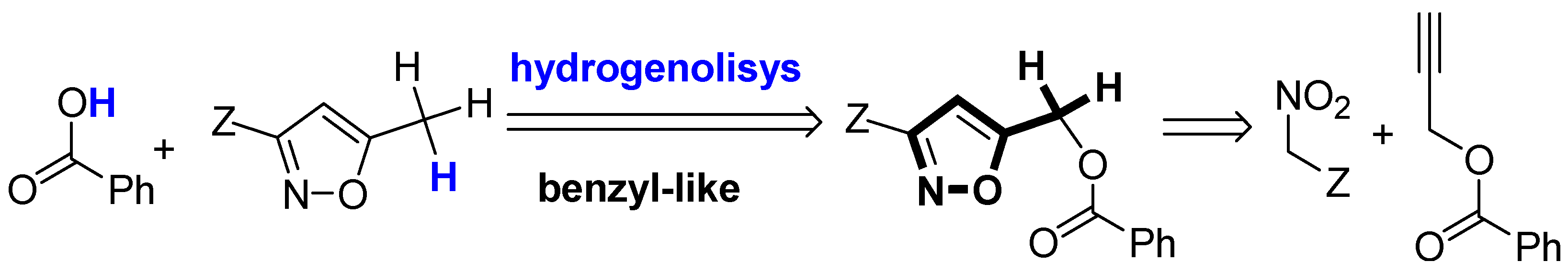


{kind=link}
{kind=link}
{kind=link}