
(1R/S,7aS/R)-1-Benzyl-1-[2,8-bis(trifluoromethyl)quinolin-4-yl]-hexahydro-oxazolo[3,4-a]pyridin-3-one


Abstract
: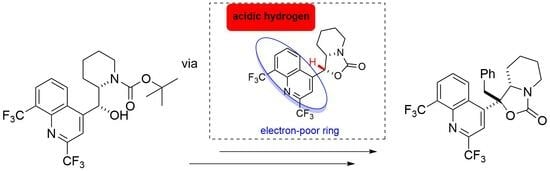
1. Introduction
2. Results and Discussion
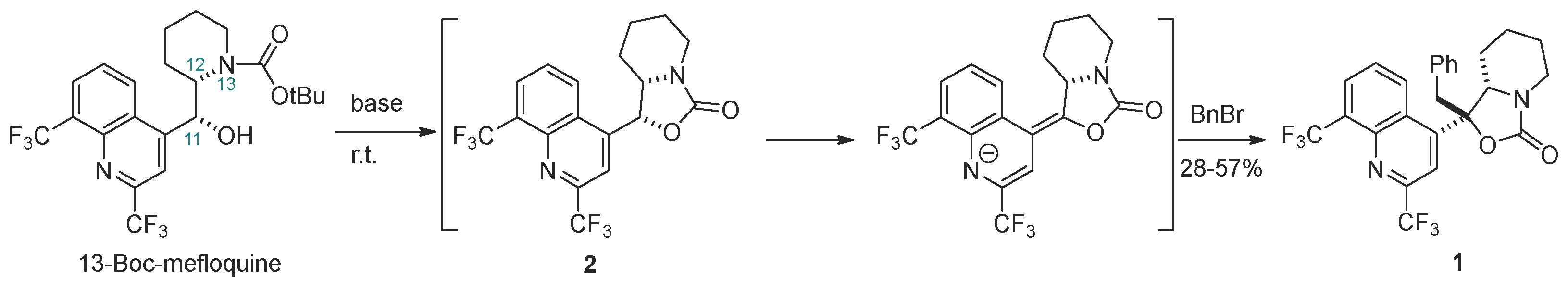
2.1. Synthesis
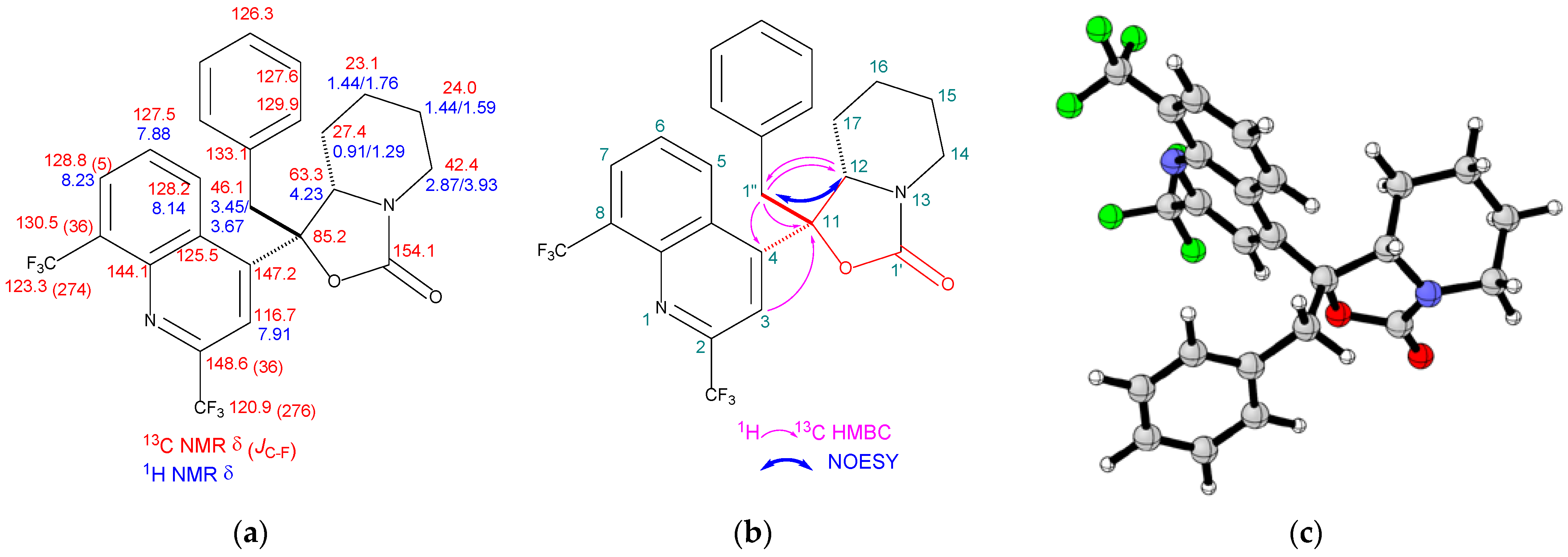
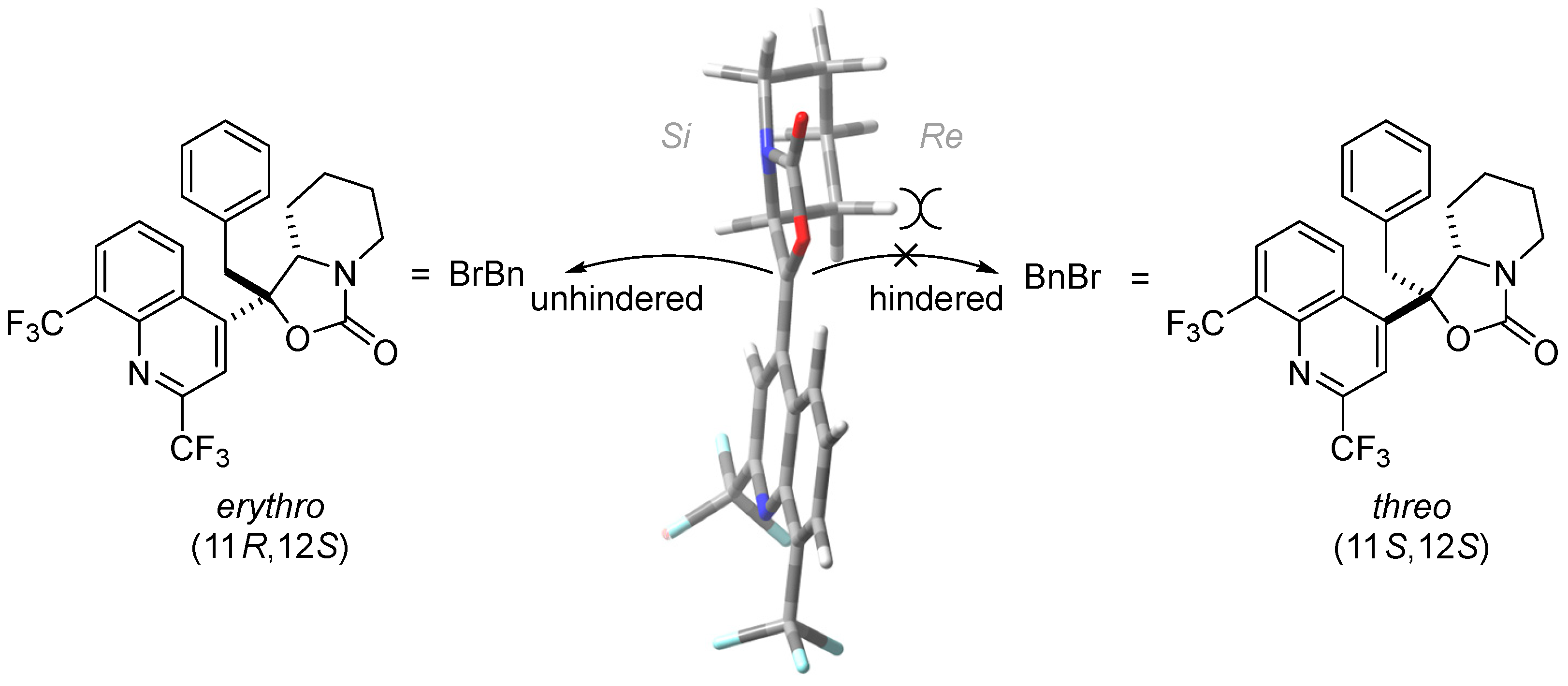
2.2. Structure Elucidation
3. Discussion
4. Materials and Methods
4.1. (1R/S,7aS/R)-1-Benzyl-1-[2,8-bis(trifluoromethyl)quinolin-4-yl]-hexahydro-oxazolo[3,4-a]pyridin-3-one (1)
4.2. (1R/S,7aS/R)-1-[2,8-Bis(trifluoromethyl)quinolin-4-yl]-hexahydro-oxazolo[3,4-a]pyridin-3-one (2) [6]
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Kucharski, D.J.; Jaszczak, M.; Boratyński, P.J. A Review of Modifications of Quinoline Antimalarials: Mefloquine and (hydroxy)Chloroquine. Molecules 2022, 27, 1003. [Google Scholar] [CrossRef] [PubMed]
- Kucharski, D.J.; Kowalczyk, R.; Boratyński, P.J. Chiral Vicinal Diamines Derived from Mefloquine. J. Org. Chem. 2021, 86, 10654–10664. [Google Scholar] [CrossRef] [PubMed]
- Kucharski, D.J.; Suchanek, R.; Kowalczyk, R.; Boratyński, P.J. Development of Mefloquine-Based Bifunctional Secondary Amine Organocatalysts for Enantioselective Michael and Friedel–Crafts Reactions. J. Org. Chem. 2023, ASAP. [Google Scholar] [CrossRef] [PubMed]
- Engwerda, A.H.J.; Maassen, R.; Tinnemans, P.; Meekes, H.; Rutjes, F.P.J.T.; Vlieg, E. Attrition-Enhanced Deracemization of the Antimalaria Drug Mefloquine. Angew. Chem. Int. Ed. 2019, 58, 1670–1673. [Google Scholar] [CrossRef] [PubMed]
- Agami, C.; Couty, F. The Reactivity of the N-Boc Protecting Group: An Underrated Feature. Tetrahedron 2002, 58, 2701–2724. [Google Scholar] [CrossRef]
- Gillespie, R.J.; Lerpiniere, J.; Giles, P.R.; Adams, D.R.; Knutsen, L.J.S.; Cliffe, I.A. 4-Quinolinemethanol Derivatives as Purine Receptor Antagonists (II). U.S. Patent 6608085 (B1), 19 August 2003. [Google Scholar]
- Havaran, L.M.; Chong, D.C.; Childers, W.E.; Dollings, P.J.; Dietrich, A.; Harrison, B.L.; Marathias, V.; Tawa, G.; Aulabaugh, A.; Cowling, R.; et al. 3,4-Dihydropyrimido(1,2-a)Indol-10(2H)-Ones as Potent Non-Peptidic Inhibitors of Caspase-3. Bioorg. Med. Chem. 2009, 17, 7755–7768. [Google Scholar] [CrossRef] [PubMed]
- Franceschini, N.; Sonnet, P.; Guillaume, D. Simple, Versatile and Highly Diastereoselective Synthesis of 1,3,4-Trisubstituted-2-Oxopiperazine-Containing Peptidomimetic Precursors. Org. Biomol. Chem. 2005, 3, 787–793. [Google Scholar] [CrossRef] [PubMed]
- El Aissi, R.; Liu, J.; Besse, S.; Canitrot, D.; Chavignon, O.; Chezal, J.-M.; Miot-Noirault, E.; Moreau, E. Synthesis and Biological Evaluation of New Quinoxaline Derivatives of ICF01012 as Melanoma-Targeting Probes. ACS Med. Chem. Lett. 2014, 5, 468–473. [Google Scholar] [CrossRef]
- Herdeis, C.; Kaschinski, C.; Karla, R.; Lotter, H. Synthesis of Homochiral Piperidine Derivatives from S-Glutamic Acid. Stereoselective 1,4-Addition of Organocuprates to a Δ3-Piperidine-2-One. A Paroxetine Analogue. Tetrahedron Asymmetry 1996, 7, 867–884. [Google Scholar] [CrossRef]
- Koenigs, W. Ueber Derivate Des Lepidins. Berichte der Dtsch. Chem. Ges. 1898, 31, 2364–2376. [Google Scholar] [CrossRef]
- Vu, V.H.; Bouvry, C.; Roisnel, T.; Golhen, S.; Hurvois, J.-P. Formal Synthesis of (–)-Perhydrohistrionicotoxin Using a Thorpe-Ziegler Cyclization Approach. Synthesis of Functionalized Aza-Spirocycles. Eur. J. Org. Chem. 2019, 2019, 1215–1224. [Google Scholar] [CrossRef]
- Osuch, C.; Levine, R. The Use of Organolithium Compounds to Effect the Alkylation of 2- and 4-Picoline1. J. Am. Chem. Soc. 1956, 78, 1723–1725. [Google Scholar] [CrossRef]
- Knight, J.D.; Sauer, S.J.; Coltart, D.M. Asymmetric Total Synthesis of the Antimalarial Drug (+)-Mefloquine Hydrochloride via Chiral N-Amino Cyclic Carbamate Hydrazones. Org. Lett. 2011, 13, 3118–3121. [Google Scholar] [CrossRef] [PubMed]
- Kansal, V.K.; Maniyan, P.P.; Deshmukh, S.S.; Gupta, N.L. A Process for the Stereospecific Synthesis of Erythro-Mefloquine Hydrochloride. India Patent IN185066B, 4 November 2000. [Google Scholar]
- Kansal, V.K.; Maniyan, P.P.; Deshmukh, S.S. An Improved Process for the Manufacture of Erythro-Mefloquine Hydrochloride. India Patent IN185394B, 13 January 2001. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Alkyl Halide | Base/Reaction Conditions | Product, Yield % |
|---|---|---|---|
| 1 | BnBr | None, r.t to 70 °C | 0 |
| 2 | BnBr | KOH, r.t. | 1, 28 |
| 3 | BnBr | NaH, r.t. | 1, 57 |
| 4 | MeI | NaH, r.t. | 2, 17 |
| 5 | MeI | KOH, r.t, | 2, 42 |
| 6 | none 1 | PPh3, DIAD, HN3, r.t. | 2, 68 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kucharski, D.J.; Boratyński, P.J. (1R/S,7aS/R)-1-Benzyl-1-[2,8-bis(trifluoromethyl)quinolin-4-yl]-hexahydro-oxazolo[3,4-a]pyridin-3-one. Molbank 2024, 2024, M1756. https://doi.org/10.3390/M1756
Kucharski DJ, Boratyński PJ. (1R/S,7aS/R)-1-Benzyl-1-[2,8-bis(trifluoromethyl)quinolin-4-yl]-hexahydro-oxazolo[3,4-a]pyridin-3-one. Molbank. 2024; 2024(1):M1756. https://doi.org/10.3390/M1756
Chicago/Turabian StyleKucharski, Dawid J., and Przemysław J. Boratyński. 2024. "(1R/S,7aS/R)-1-Benzyl-1-[2,8-bis(trifluoromethyl)quinolin-4-yl]-hexahydro-oxazolo[3,4-a]pyridin-3-one" Molbank 2024, no. 1: M1756. https://doi.org/10.3390/M1756
APA StyleKucharski, D. J., & Boratyński, P. J. (2024). (1R/S,7aS/R)-1-Benzyl-1-[2,8-bis(trifluoromethyl)quinolin-4-yl]-hexahydro-oxazolo[3,4-a]pyridin-3-one. Molbank, 2024(1), M1756. https://doi.org/10.3390/M1756