
4-(5-Benzyl-3-((4-fluorophenyl)sulfonyl)-5-methyl-4,5-dihydrofuran-2-yl)-2-nitrobenzamide

 , , , and
, , , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
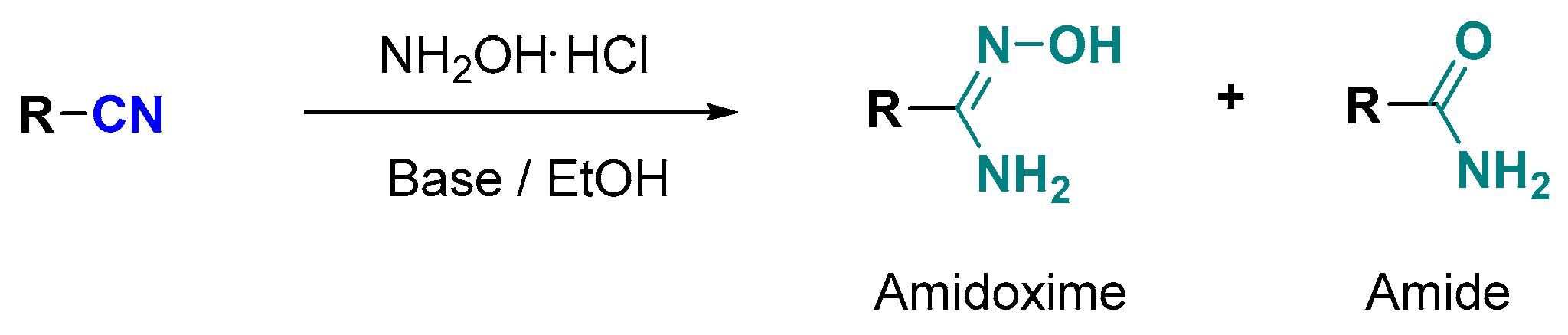
:1. Introduction
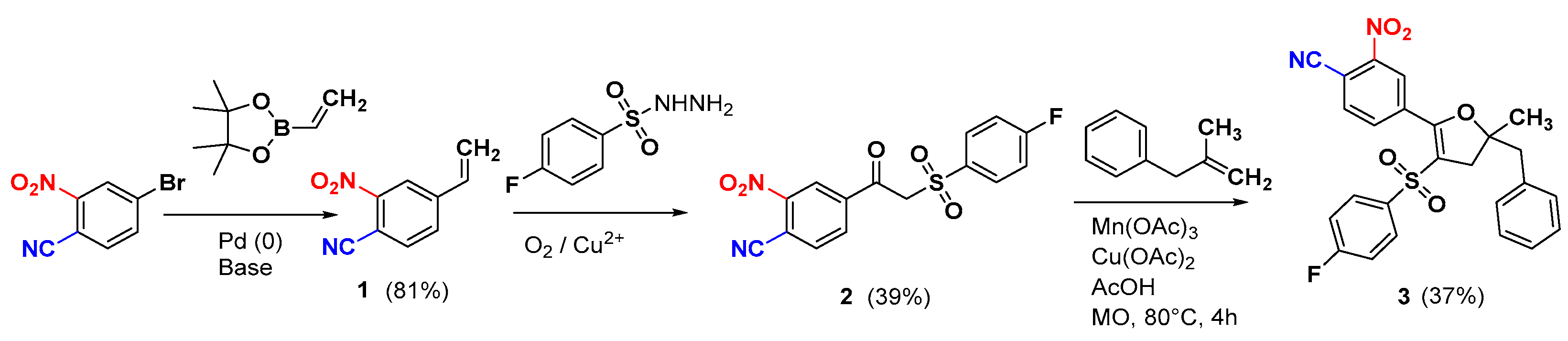
2. Results
3. Materials and Methods
3.1. Chemistry
3.2. 2-Nitro-4-vinylbenzonitrile (1)
3.3. 4-(2-((4-Fluorophenyl)sulfonyl)acetyl)-2-nitrobenzonitrile (2)
3.4. 4-(5-Benzyl-3-((4-fluorophenyl)sulfonyl)-5-methyl-4,5-dihydrofuran-2-yl)-2-nitrobenzonitrile (3)
3.5. 4-(5-Benzyl-3-((4-fluorophenyl)sulfonyl)-5-methyl-4,5-dihydrofuran-2-yl)-2-nitrobenzamide (4)
3.6. 4-(5-Benzyl-3-((4-fluorophenyl)sulfonyl)-5-methyl-4,5-dihydrofuran-2-yl)-N’-hydroxy-2-nitrobenzimidamide (5)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yang, H.; Liu, J.; Wu, W.; Luo, Y.; Mo, G.; Huang, K. Synthesis of Amidoxime-modified Hollow Organic Porous Nanospheres for Efficient Uranium Adsorption from Aqueous Solution. ChemistrySelect 2023, 8, e202301214. [Google Scholar] [CrossRef]
- Tang, J.; Bai, X.; Huang, H.; Xue, S.; Pan, J. Templating Synthesis of Oxime/Amidoxime Functionalized Hollow Nanospheres by Air Bubbles Generated from “Ouzo-Like” Effect for Fast and Massive Uranium Uptake. Sep. Purif. Technol. 2023, 306, 122463. [Google Scholar] [CrossRef]
- Ahmad, Z.; Li, Y.; Yang, J.; Geng, N.; Fan, Y.; Gou, X.; Sun, Q.; Chen, J. A Membrane-Supported Bifunctional Poly(Amidoxime-ethyleneimine) Network for Enhanced Uranium Extraction from Seawater and Wastewater. J. Hazard. Mater. 2022, 425, 127995. [Google Scholar] [CrossRef] [PubMed]
- Lutsenko, I.A.; Vologzhanina, A.V.; Kayukova, L.A.; Yergalieva, E.M.; Koshenskova, K.A.; Bekker, O.B.; Dorovatovskii, P.V.; Eremenko, I.L. Mixed-Valence Hexanuclear CoII,III Complex with Amidoxime: Synthesis, Structure, and in Vitro Biological Activity against the Non-Pathogenic Strain of Mycolicibacterium Smegmatis. Russ. Chem. Bull. 2022, 71, 2172–2178. [Google Scholar] [CrossRef]
- Bhavsar, S.; Tadiparthi, R.; Pawar, S.; Kayastha, A.K.; Chavan, V.; Yeole, R.; Deshpande, P.; Bhagwat, S.; Patel, M. Design of Novel Amidoxime Ketolide Core and an Efficient Synthesis of WCK 4763: For Treatment of Gram-Positive Pneumococci. Med. Chem. Res. 2023, 32, 1726–1735. [Google Scholar] [CrossRef]
- Berger, O.; Ortial, S.; Wein, S.; Denoyelle, S.; Bressolle, F.; Durand, T.; Escale, R.; Vial, H.J.; Vo-Hoang, Y. Evaluation of Amidoxime Derivatives as Prodrug Candidates of Potent Bis-Cationic Antimalarials. Bioorganic Med. Chem. Lett. 2019, 29, 2203–2207. [Google Scholar] [CrossRef] [PubMed]
- Sahyoun, T.; Arrault, A.; Schneider, R. Amidoximes and Oximes: Synthesis, Structure, and Their Key Role as NO Donors. Molecules 2019, 24, 2470. [Google Scholar] [CrossRef] [PubMed]
- Fylaktakidou, K.; Hadjipavlou-Litina, D.; Litinas, K.; Varella, E.; Nicolaides, D. Recent Developments in the Chemistry and in the Biological Applications of Amidoximes. CPD 2008, 14, 1001–1047. [Google Scholar] [CrossRef]
- Tabélé, C.; Faiões, V.D.S.; Grimaud, F.; Torres-Santos, E.C.; Khoumeri, O.; Curti, C.; Vanelle, P. Original Antileishmanial Hits: Variations around Amidoximes. Eur. J. Med. Chem. 2018, 148, 154–164. [Google Scholar] [CrossRef]
- Avendaño Leon, O.L.; Curti, C.; Santos Urbancg Moncorvo, F.M.; Kabri, Y.; Redon, S.; Torres-Santos, E.C.; Paoli-Lombardo, R.; Vanelle, P. Diethyl (5-Benzyl-2-(4-(N′-Hydroxycarbamimidoyl)Phenyl)-5-Methyl-4,5-Dihydrofuran-3-Yl)Phosphonate. Molbank 2023, 2023, M1736. [Google Scholar] [CrossRef]
- Festa, C.; Finamore, C.; Marchianò, S.; Di Leva, F.S.; Carino, A.; Monti, M.C.; Del Gaudio, F.; Ceccacci, S.; Limongelli, V.; Zampella, A.; et al. Investigation around the Oxadiazole Core in the Discovery of a New Chemotype of Potent and Selective FXR Antagonists. ACS Med. Chem. Lett. 2019, 10, 504–510. [Google Scholar] [CrossRef] [PubMed]
- Tang, N.; Liang, J.; Niu, C.; Wang, H.; Luo, Y.; Xing, W.; Ye, S.; Liang, C.; Guo, H.; Guo, J.; et al. Amidoxime-Based Materials for Uranium Recovery and Removal. J. Mater. Chem. A 2020, 8, 7588–7625. [Google Scholar] [CrossRef]
- Faïon, L.; Djaout, K.; Pintiala, C.; Piveteau, C.; Leroux, F.; Biela, A.; Slupek, S.; Antoine, R.; Záhorszká, M.; Cantrelle, F.-X.; et al. Exploring the Antitubercular Activity of Anthranilic Acid Derivatives: From MabA (FabG1) Inhibition to Intrabacterial Acidification. Pharmaceuticals 2023, 16, 335. [Google Scholar] [CrossRef] [PubMed]
- Shabalin, D.A.; Dunsford, J.J.; Ngwerume, S.; Saunders, A.R.; Gill, D.M.; Camp, J.E. Synthesis of 2,4-Disubstituted Imidazoles via Nucleophilic Catalysis. Synlett 2020, 31, 797–800. [Google Scholar] [CrossRef]
- Murarka, S.; Martín-Gago, P.; Schultz-Fademrecht, C.; Al Saabi, A.; Baumann, M.; Fansa, E.K.; Ismail, S.; Nussbaumer, P.; Wittinghofer, A.; Waldmann, H. Development of Pyridazinone Chemotypes Targeting the PDEδ Prenyl Binding Site. Chem. Eur. J. 2017, 23, 6083–6093. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.N.; Bhowmick, R.R.; Nurnabi, M.; Molla, A.I. Synthesis and Characterization of an Amidoxime Compound with Benzimidazole Moiety and pH Sensing Behavior Under Basic Condition in Methanol and DMSO. J. Chil. Chem. Soc. 2018, 63, 4047–4050. [Google Scholar] [CrossRef]
- Bouhlel, A.; Curti, C.; Dumètre, A.; Laget, M.; Crozet, M.D.; Azas, N.; Vanelle, P. Synthesis and Evaluation of Original Amidoximes as Antileishmanial Agents. Bioorg. Med. Chem. 2010, 18, 7310–7320. [Google Scholar] [CrossRef]
- Bakunova, S.M.; Bakunov, S.A.; Wenzler, T.; Barszcz, T.; Werbovetz, K.A.; Brun, R.; Tidwell, R.R. Synthesis and Antiprotozoal Activity of Pyridyl Analogues of Pentamidine. J. Med. Chem. 2009, 52, 4657–4667. [Google Scholar] [CrossRef]
- Kirby, A.J.; Davies, J.E.; Brandão, T.A.S.; Da Silva, P.F.; Rocha, W.R.; Nome, F. Hydroxylamine as an Oxygen Nucleophile. Structure and Reactivity of Ammonia Oxide. J. Am. Chem. Soc. 2006, 128, 12374–12375. [Google Scholar] [CrossRef]
- Medeiros, M.; Souza, B.S.; Orth, E.S.; Brandão, T.A.S.; Rocha, W.; Kirby, A.J.; Nome, F. The Reaction of Hydroxylamine with Aspirin. Arkivoc 2011, 2011, 461–476. [Google Scholar] [CrossRef]
- Buncel, E.; Um, I.H.; Terrier, F. Hydroxylamine, Oximate and Hydroxamate as α-Nucleophiles in Dephosphorylation. In PATAI’S Chemistry of Functional Groups; Rappoport, Z., Liebman, J.F., Eds.; Wiley: New York, NY, USA, 2008; pp. 817–837. ISBN 978-0-470-51261-6. [Google Scholar]
- Srivastava, R.M.; Pereira, M.C.; Faustino, W.W.M.; Coutinho, K.; Dos Anjos, J.V.; De Melo, S.J. Synthesis, Mechanism of Formation, and Molecular Orbital Calculations of Arylamidoximes. Monatsh. Chem. 2009, 140, 1319–1324. [Google Scholar] [CrossRef]
- Stephenson, L.; Warburton, W.K.; Wilson, M.J. Reaction of Some Aromatic Nitriles with Hydroxylamine to Give Amides, and an Alternative Preparation of Amidoximes. J. Chem. Soc. C 1969, 6, 861. [Google Scholar] [CrossRef]
- Voros, A.; Baan, Z.; Timari, G.; Hermecz, I.; Mizsey, P.; Finta, Z. Highly Efficient and Selective Addition of Hydroxylamine to Nitriles in Ionic Liquids. COS 2014, 11, 751–756. [Google Scholar] [CrossRef]
- Vörös, A.; Mucsi, Z.; Baán, Z.; Timári, G.; Hermecz, I.; Mizsey, P.; Finta, Z. An Experimental and Theoretical Study of Reaction Mechanisms between Nitriles and Hydroxylamine. Org. Biomol. Chem. 2014, 12, 8036–8047. [Google Scholar] [CrossRef] [PubMed]
- LoPachin, R.M.; Geohagen, B.C.; Nordstroem, L.U. Mechanisms of Soft and Hard Electrophile Toxicities. Toxicology 2019, 418, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Pearson, R.G. Hard and Soft Acids and Bases, HSAB, Part 1: Fundamental Principles. J. Chem. Educ. 1968, 45, 581. [Google Scholar] [CrossRef]
- Meredith, N.Y.; Borsley, S.; Smolyar, I.V.; Nichol, G.S.; Baker, C.M.; Ling, K.B.; Cockroft, S.L. Dissecting Solvent Effects on Hydrogen Bonding. Angew. Chem. Int. Ed. 2022, 61, e202206604. [Google Scholar] [CrossRef]
- Graneek, J.B.; Bailey, W.C.; Schnell, M. Electron-Withdrawing Effects on the Molecular Structure of 2- and 3-Nitrobenzonitrile Revealed by Broadband Rotational Spectroscopy and Their Comparison with 4-Nitrobenzonitrile. Phys. Chem. Chem. Phys. 2018, 20, 22210–22217. [Google Scholar] [CrossRef]
- Jezuita, A.; Ejsmont, K.; Szatylowicz, H. Substituent Effects of Nitro Group in Cyclic Compounds. Struct. Chem. 2021, 32, 179–203. [Google Scholar] [CrossRef]
- Chambers, R.D.; Edwards, A.R.; Batsanov, A.; Howard, J.A.K. Ambident Nucleophilic Addition to Ethyl Trifluoromethylacetoacetate. J. Fluor. Chem. 1998, 88, 95–97. [Google Scholar] [CrossRef]
- Fernández, M.I.; Canle, M.; García, M.V.; Santaballa, J.A. A Theoretical Analysis of the Acid–Base Equilibria of Hydroxylamine in Aqueous Solution. Chem. Phys. Lett. 2010, 490, 159–164. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Avendaño Leon, O.L.; Curti, C.; El-Kashef, H.; Kabri, Y.; Redon, S.; Vanelle, P. 4-(5-Benzyl-3-((4-fluorophenyl)sulfonyl)-5-methyl-4,5-dihydrofuran-2-yl)-2-nitrobenzamide. Molbank 2023, 2023, M1750. https://doi.org/10.3390/M1750
Avendaño Leon OL, Curti C, El-Kashef H, Kabri Y, Redon S, Vanelle P. 4-(5-Benzyl-3-((4-fluorophenyl)sulfonyl)-5-methyl-4,5-dihydrofuran-2-yl)-2-nitrobenzamide. Molbank. 2023; 2023(4):M1750. https://doi.org/10.3390/M1750
Chicago/Turabian StyleAvendaño Leon, Oscar Leonardo, Christophe Curti, Hussein El-Kashef, Youssef Kabri, Sébastien Redon, and Patrice Vanelle. 2023. "4-(5-Benzyl-3-((4-fluorophenyl)sulfonyl)-5-methyl-4,5-dihydrofuran-2-yl)-2-nitrobenzamide" Molbank 2023, no. 4: M1750. https://doi.org/10.3390/M1750
APA StyleAvendaño Leon, O. L., Curti, C., El-Kashef, H., Kabri, Y., Redon, S., & Vanelle, P. (2023). 4-(5-Benzyl-3-((4-fluorophenyl)sulfonyl)-5-methyl-4,5-dihydrofuran-2-yl)-2-nitrobenzamide. Molbank, 2023(4), M1750. https://doi.org/10.3390/M1750