
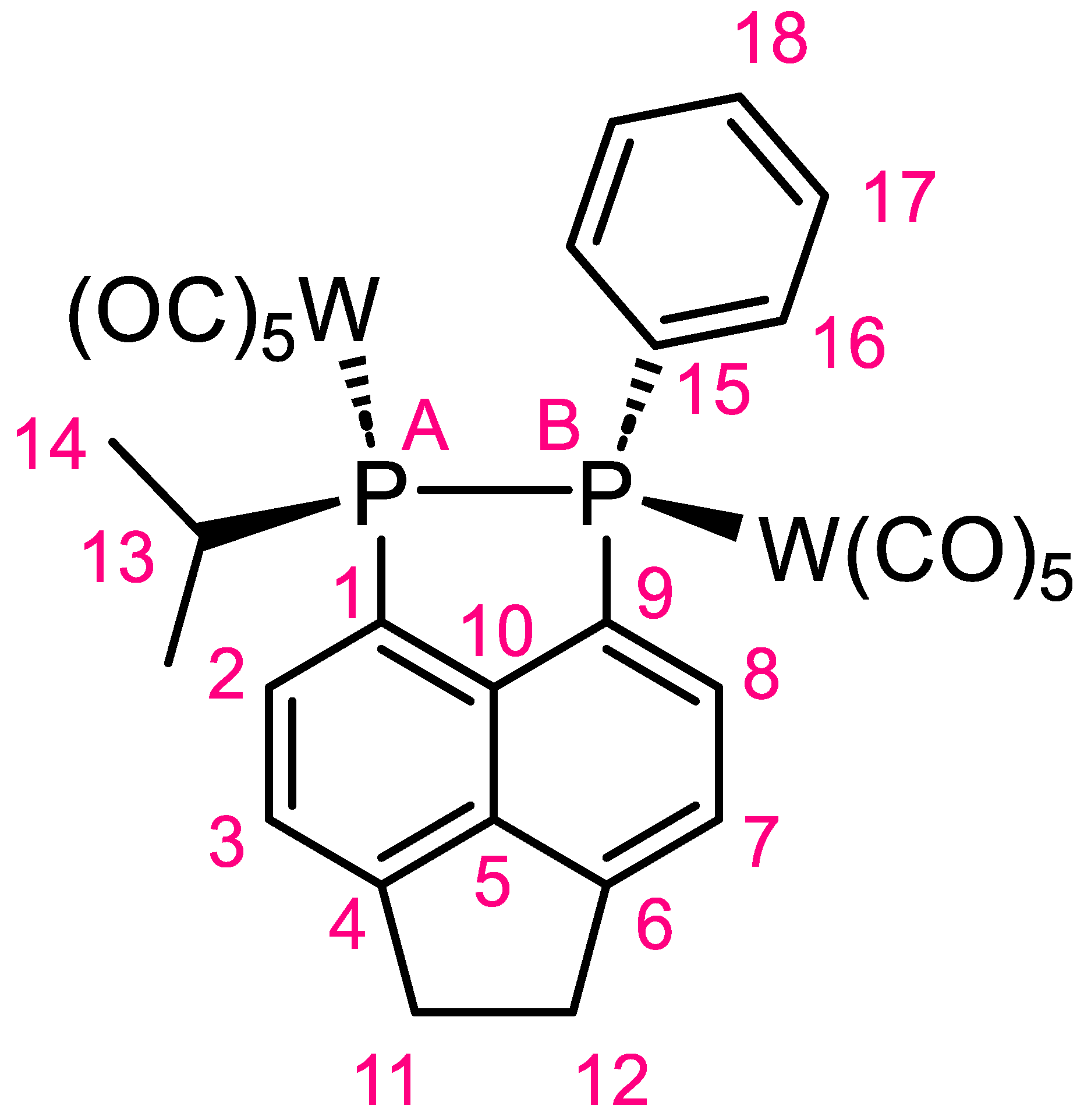
(Decacarbonyl)(1-isopropyl-2-phenyl-1,2,5,6-tetrahydroacenaphtho [5,6-cd][1,2]diphosphole)ditungsten(0)

 , , , and
, , , and
Abstract
1. Introduction
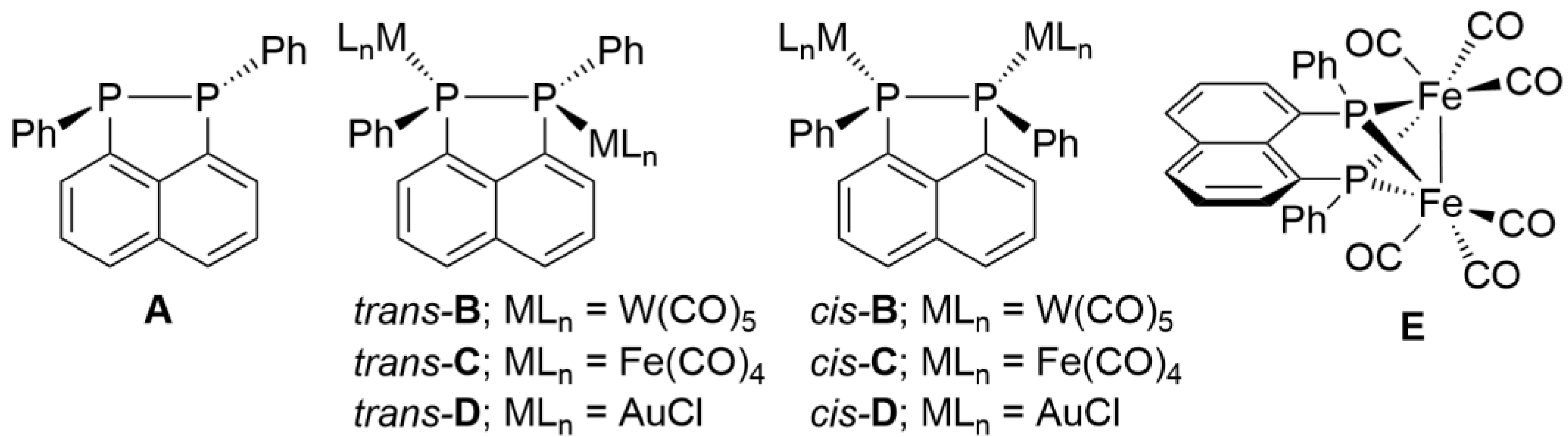
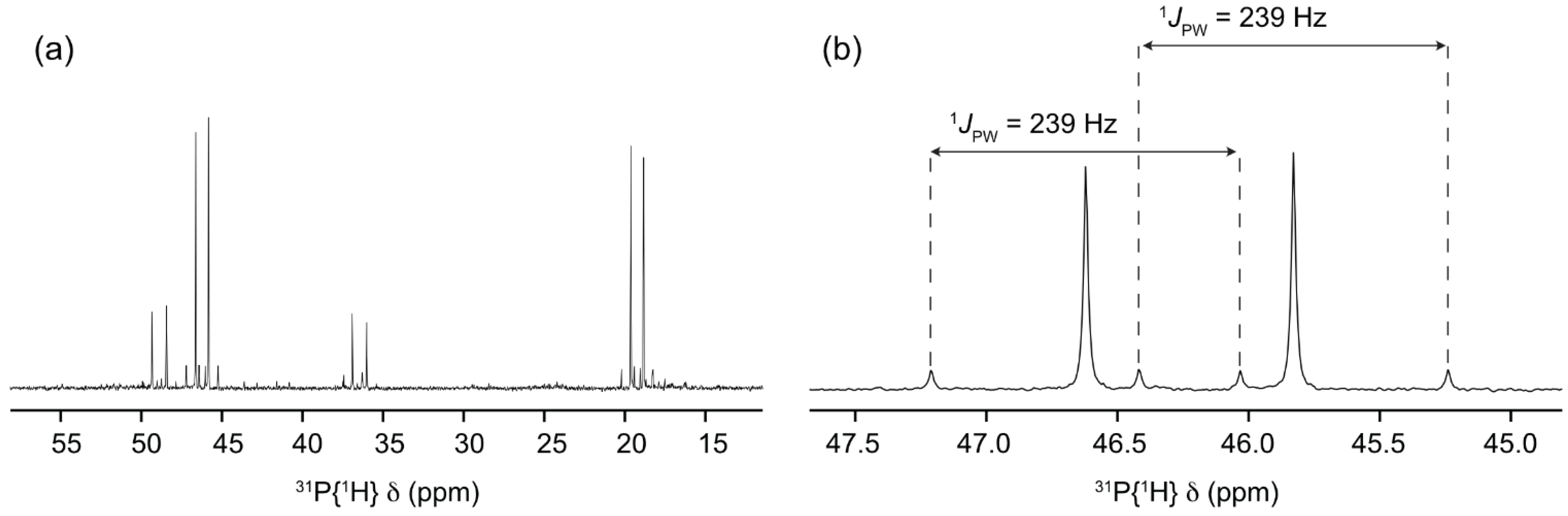
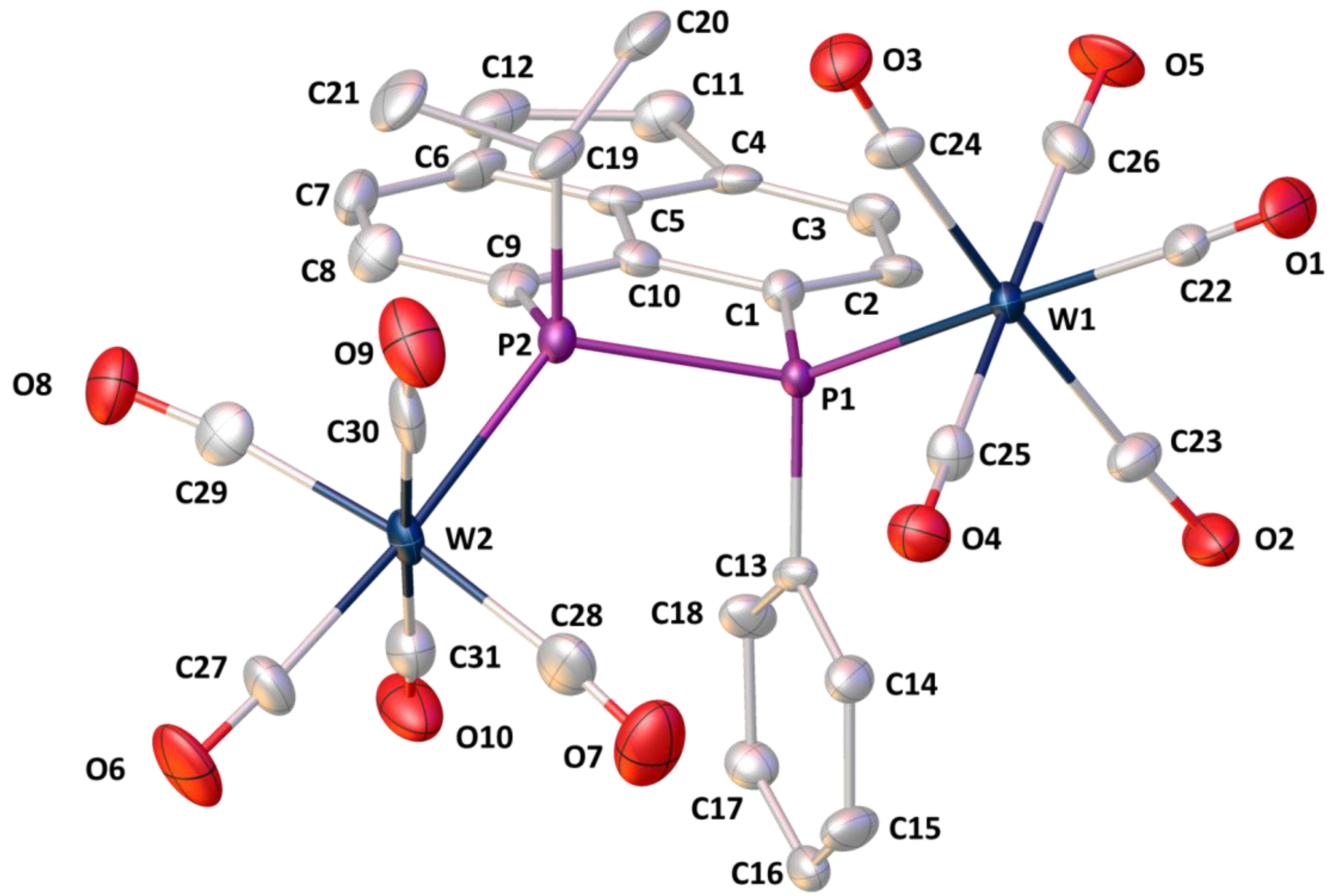
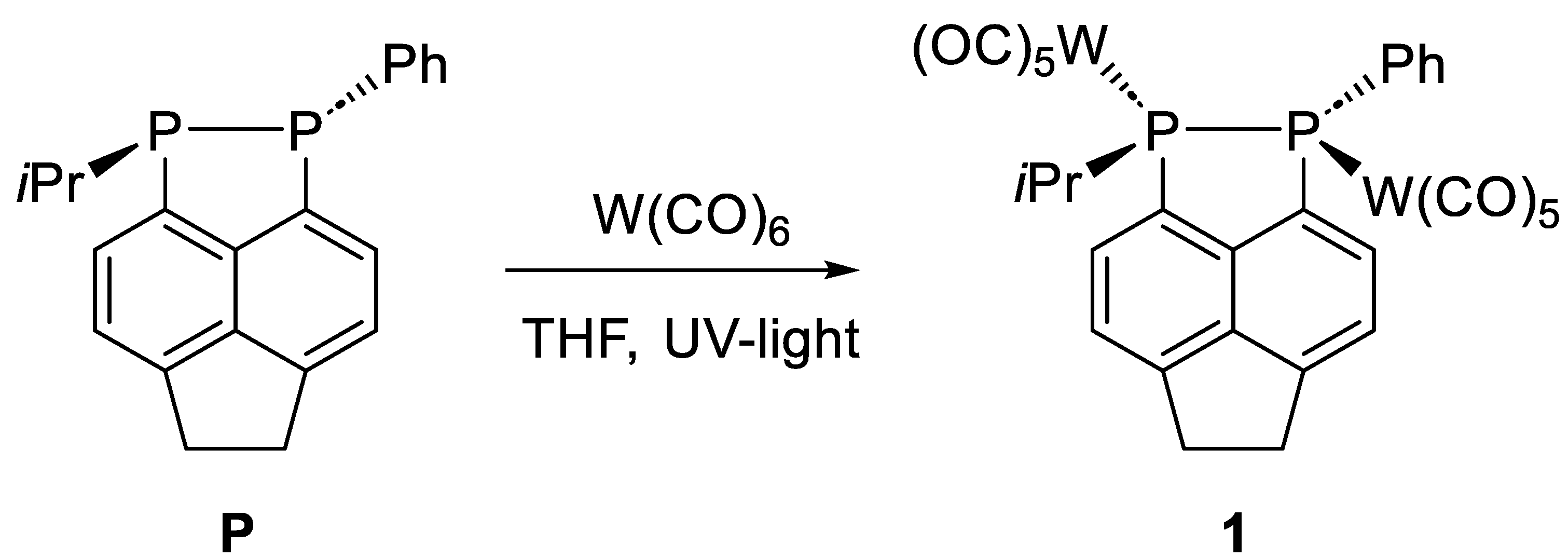
2. Results
3. Materials and Methods
3.1. General Considerations
3.2. Synthetic Procedure
Synthesis of (Decacarbonyl)(1-isopropyl-2-phenyl-1,2,5,6-tetrahydroacenaphtho [5,6-cd][1,2]diphosphole)ditungsten(0) (1)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mizuta, T.; Kunikata, S.; Miyoshi, K. Synthesis and molecular structures of dinuclear complexes with 1,2-dihydro-1,2-diphenyl-naphtho[1,8-c,d]1,2-diphosphole as a bridging ligand. J. Organomet. Chem. 2004, 689, 2624–2632. [Google Scholar] [CrossRef]
- Teramoto, Y.; Kubo, K.; Kume, S.; Mizuta, T. Formation of a Hexacarbonyl Diiron Complex Having a Naphthalene-1,8-bis(phenylphosphido) Bridge and the Electrochemical Behavior of Its Derivatives. Organometallics 2013, 32, 7014–7024. [Google Scholar] [CrossRef]
- Teramoto, Y.; Kubo, K.; Mizuta, T. PhP–PPh group bound to 1,8-positions of naphthalene: Preparation of cis isomer and synthesis of binuclear complex. J. Organomet. Chem. 2011, 696, 3402–3407. [Google Scholar] [CrossRef]
- Taylor, L.J.; Bühl, M.; Chalmers, B.A.; Ray, M.J.; Wawrzyniak, P.; Walton, J.C.; Cordes, D.B.; Slawin, A.M.Z.; Woollins, J.D.; Kilian, P. Dealkanative Main Group Couplings across the peri-Gap. J. Am. Chem. Soc. 2017, 139, 18545–18551. [Google Scholar] [CrossRef] [PubMed]
- Corbridge, Derek, E.C. Phosphorus World: Chemistry, Biochemistry and Technology; CD-ROM; Great Britain: Harrogate, UK, 2005; p. 76. [Google Scholar]
- Thomas, I.R.; Bruno, I.L.; Cole, J.C.; Macrae, C.F.; Pidcock, E.; Wood, P.A. WebCSD: The online portal to the Cambridge Structural Database. J. Appl. Cryst. 2010, 43, 362–366. [Google Scholar] [CrossRef] [PubMed]
- Fuller, A.L.; Scott-Hayward, L.A.S.; Li, Y.; Buhl, M.; Slawin, A.M.Z.; Woollins, J.D. Automated Chemical Crystallography. J. Am. Chem. Soc. 2010, 132, 5799–5802. [Google Scholar] [CrossRef] [PubMed]
- CrystalClear-SM Expert v2.1, Rigaku Americas: The Woodlands, TX, USA; Rigaku Corporation: Tokyo, Japan, 2015.
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- CrystalStructure v4.3.0, Rigaku Americas: The Woodlands, TX, USA; Rigaku Corporation: Tokyo, Japan, 2018.
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peri-Region Bond Lengths | Peri-Region Bond Angles | ||
|---|---|---|---|
| P1−P2 | 2.261(6) | CPh−P1−W1 | 115.3(5) |
| P1−W1 | 2.532(3) | CiPr−P2−W2 | 117.2(5) |
| P2−W2 | 2.501(4) | P1−P2−W2 | 120.7(2) |
| W1···W2 | 5.856(1) | P2−P1−W1 | 123.6(2) |
| Splay angle (a) | −8(1) | ||
| Out-of-plane displacements (b) | Dihedral angles | ||
| P1 | 0.098 | P1−C1···C9−P2 | 10.1(6) |
| P2 | 0.197 | C1−C10···C5−C6 | 177(1) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Davis, Z.H.; Carpenter-Warren, C.L.; Taylor, L.J.; Slawin, A.M.Z.; Kilian, P.; Chalmers, B.A. (Decacarbonyl)(1-isopropyl-2-phenyl-1,2,5,6-tetrahydroacenaphtho [5,6-cd][1,2]diphosphole)ditungsten(0). Molbank 2023, 2023, M1671. https://doi.org/10.3390/M1671
Davis ZH, Carpenter-Warren CL, Taylor LJ, Slawin AMZ, Kilian P, Chalmers BA. (Decacarbonyl)(1-isopropyl-2-phenyl-1,2,5,6-tetrahydroacenaphtho [5,6-cd][1,2]diphosphole)ditungsten(0). Molbank. 2023; 2023(2):M1671. https://doi.org/10.3390/M1671
Chicago/Turabian StyleDavis, Zachary H., Cameron L. Carpenter-Warren, Laurence J. Taylor, Alexandra M. Z. Slawin, Petr Kilian, and Brian A. Chalmers. 2023. "(Decacarbonyl)(1-isopropyl-2-phenyl-1,2,5,6-tetrahydroacenaphtho [5,6-cd][1,2]diphosphole)ditungsten(0)" Molbank 2023, no. 2: M1671. https://doi.org/10.3390/M1671
APA StyleDavis, Z. H., Carpenter-Warren, C. L., Taylor, L. J., Slawin, A. M. Z., Kilian, P., & Chalmers, B. A. (2023). (Decacarbonyl)(1-isopropyl-2-phenyl-1,2,5,6-tetrahydroacenaphtho [5,6-cd][1,2]diphosphole)ditungsten(0). Molbank, 2023(2), M1671. https://doi.org/10.3390/M1671