
Synthesis, Characterization and Chemistry of Tetrakis(Propargylisocyanide) Copper(I) Complex


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results and Discussion
3. Materials and Methods
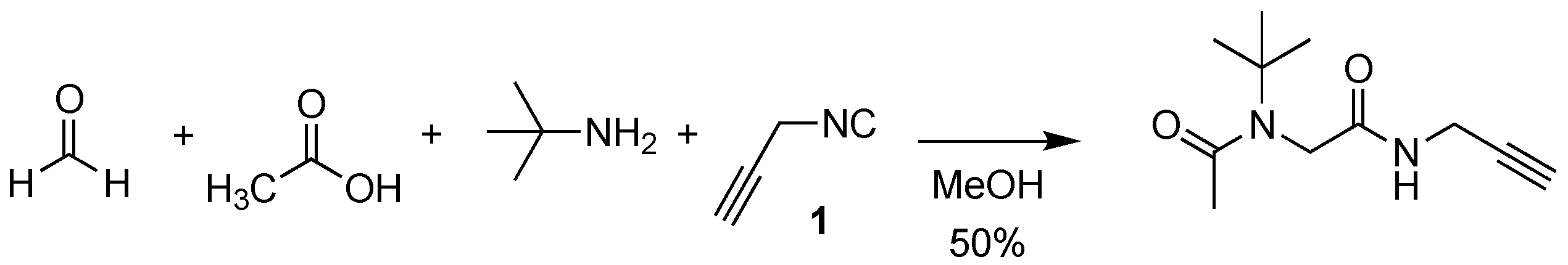
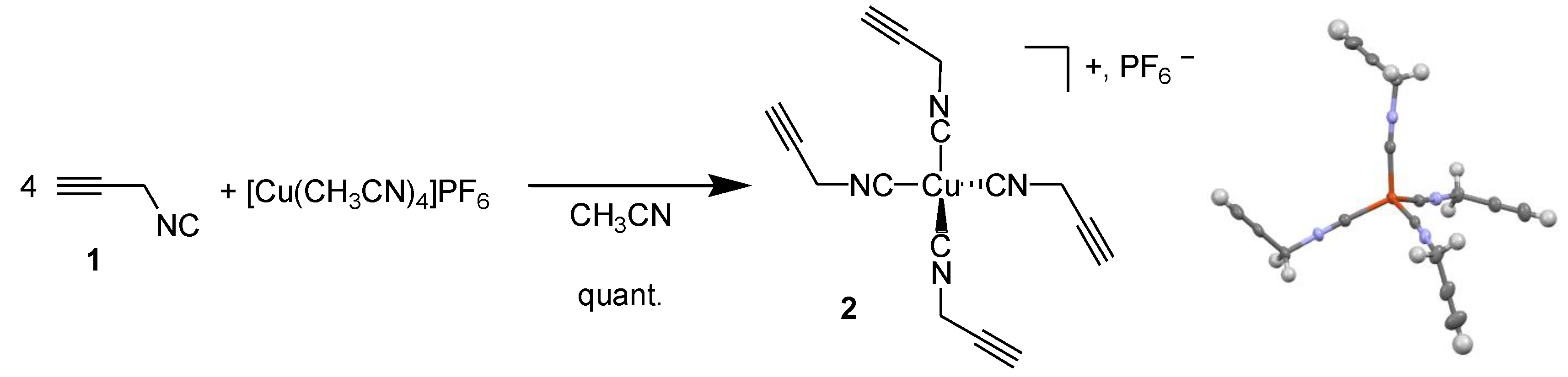
3.1. Tetrakis(propargylisonitrile) Copper(I) Hexafluorophosphate 2
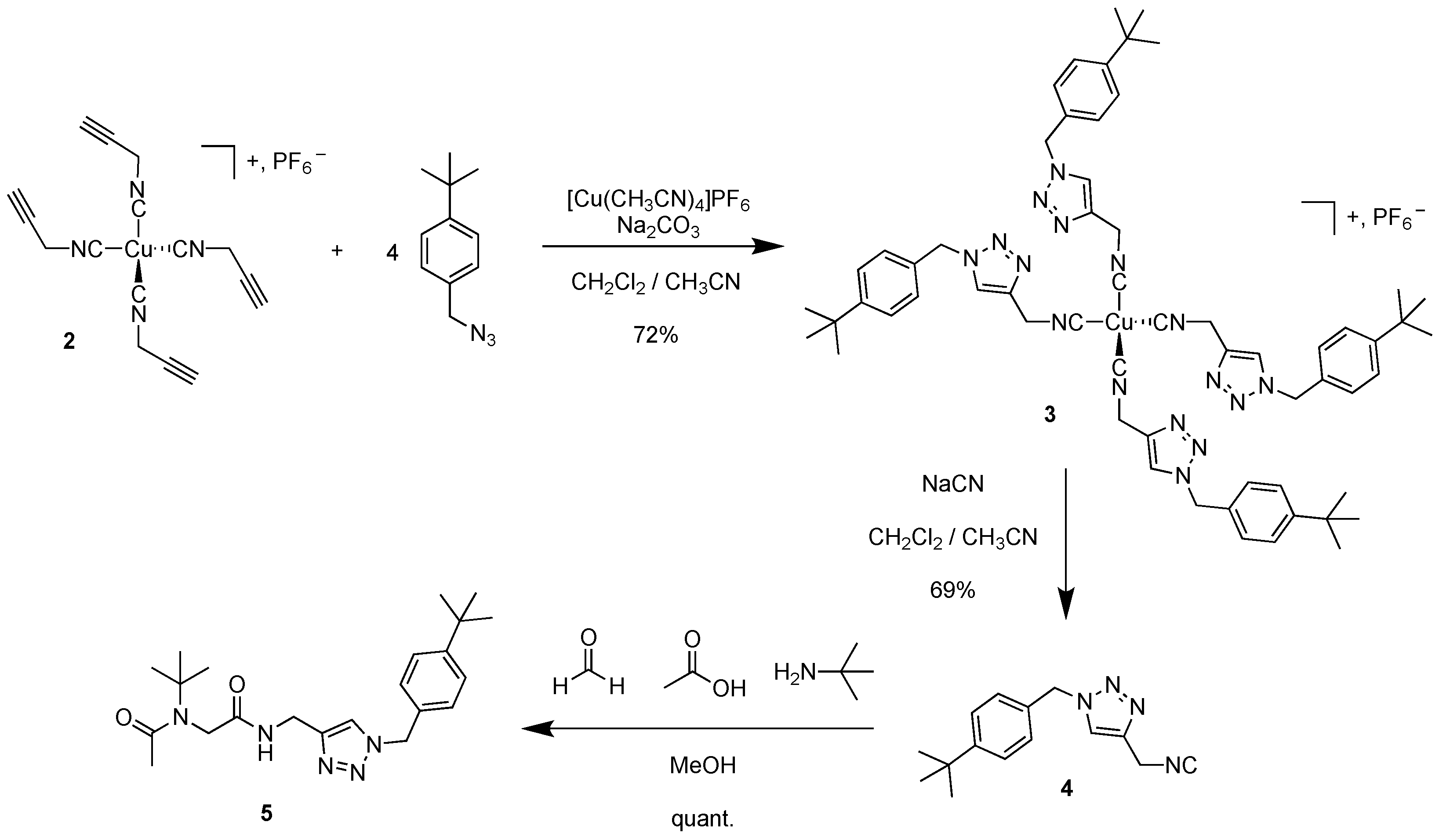
3.2. Tetrakis(1-(4-(tert-butyl)benzyl)-4-(isocyanomethyl)-1H-1,2,3-triazole) Copper(I) Hexafluorophosphate 3
3.3. 1-(4-(tert-Butyl)benzyl)-4-(isocyanomethyl)-1H-1,2,3-triazole 4
3.4. N-(tert-butyl)-N-(2-(((1-(4-(tert-butyl)benzyl)-1H-1,2,3-triazol-4-yl)methyl)amino)-2-oxoethyl)acetamide 5
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nenajdenko, V.G. Isocyanide Chemistry: Applications in Synthesis and Material Science; Wiley-VCH: Weinheim, Germany, 2012. [Google Scholar] [CrossRef]
- Van Berkel, S.S.; Boegels, B.G.M.; Wijdeven, M.A.; Westermann, B.; Rutjes, F.P.J.T. Recent Advances in Asymmetric Isocyanide-Based Multicomponent Reactions. Eur. J. Org. Chem. 2012, 2012, 3543–3559. [Google Scholar] [CrossRef]
- Wahby, Y.; Abdel-Hamid, H.; Salah Ayoup, M. Two decades of recent advances of Passerini reactions: Synthetic and potential pharmaceutical applications. New J. Chem. 2022, 45, 1445–1468. [Google Scholar] [CrossRef]
- Ugi, I.; Lohberger, S.; Karl, R. Comprehensive Organic Synthesis; Trost, B.M., Fleming, I., Eds.; Pergamon: Oxford, UK, 1991; Volume 2, p. 1083. [Google Scholar]
- Domling, A.; Huang, Y. Piperazine scaffolds via isocyanide-based multicomponent reactions. Synthesis 2010, 2010, 2859–2883. [Google Scholar] [CrossRef]
- Sadjadi, S.; Heravi, M.M. Recent application of isocyanides in synthesis of heterocycles. Tetrahedron 2011, 67, 2707–2752. [Google Scholar] [CrossRef]
- Quelhas, A.; Gazzeh, H.; Roisnel, T.; Trolez, Y.; Guillemin, J.-C. Passerini and Ugi Reactions Involving Kinetically Unstable Isocyanides. Eur. J. Org. Chem. 2021, 2021, 6002–6005. [Google Scholar] [CrossRef]
- Zwikker, J.W.; Stephany, R.W. Propargyl and Allenyl Isocyanide and Some of Their Copper(I)Complexes. Synth. Commun. 1973, 3, 19–23. [Google Scholar] [CrossRef]
- Tornøe, C.W.; Christensen, C.; Meldal, M. Peptidotriazoles on Solid Phase: [1,2,3]-Triazoles by Regiospecific Copper(I)-Catalyzed 1,3-Dipolar Cycloadditions of Terminal Alkynes to Azides. J. Org. Chem. 2002, 67, 3057–3064. [Google Scholar] [CrossRef] [PubMed]
- Rostovtsev, V.V.; Green, L.G.; Fokin, V.V.; Sharpless, K.B. A Stepwise Huisgen Cycloaddition Process: Copper(I)-Catalyzed Regioselective “Ligation” of Azides and Terminal Alkynes. Angew. Chem. Int. Ed. 2002, 41, 2596–2599. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Cryst. 2015, A71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Alvarez, S.G.; Alvarez, M.T. A Practical Procedure for the Synthesis of Alkyl Azides at Ambient Temperature in Dimethyl Sulfoxide in High Purity and Yield. Synthesis 1997, 1997, 413–414. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Quelhas, A.; Roisnel, T.; Guillemin, J.-C.; Trolez, Y. Synthesis, Characterization and Chemistry of Tetrakis(Propargylisocyanide) Copper(I) Complex. Molbank 2023, 2023, M1599. https://doi.org/10.3390/M1599
Quelhas A, Roisnel T, Guillemin J-C, Trolez Y. Synthesis, Characterization and Chemistry of Tetrakis(Propargylisocyanide) Copper(I) Complex. Molbank. 2023; 2023(1):M1599. https://doi.org/10.3390/M1599
Chicago/Turabian StyleQuelhas, Alexandre, Thierry Roisnel, Jean-Claude Guillemin, and Yann Trolez. 2023. "Synthesis, Characterization and Chemistry of Tetrakis(Propargylisocyanide) Copper(I) Complex" Molbank 2023, no. 1: M1599. https://doi.org/10.3390/M1599
APA StyleQuelhas, A., Roisnel, T., Guillemin, J.-C., & Trolez, Y. (2023). Synthesis, Characterization and Chemistry of Tetrakis(Propargylisocyanide) Copper(I) Complex. Molbank, 2023(1), M1599. https://doi.org/10.3390/M1599