

Obtainment of Threo and Erythro Isomers of the 6-Fluoro-3-(2,3,6,7,8,9-hexahydronaphtho[2,3-b][1,4]dioxin-2-yl)-2,3-dihydrobenzo[b][1,4]dioxine-5-carboxamide


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
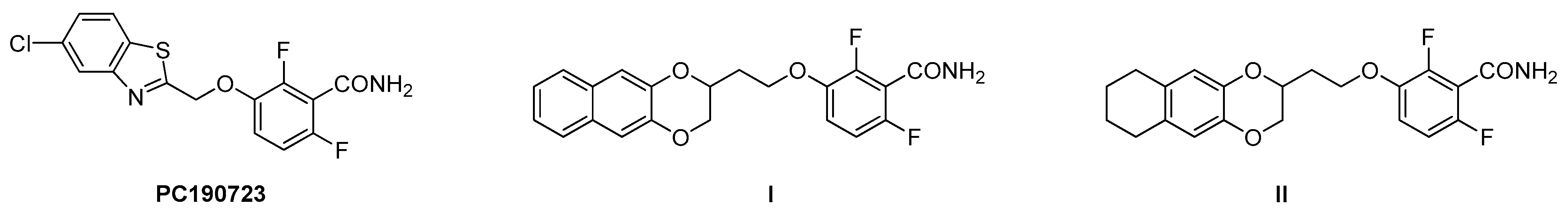
1. Introduction
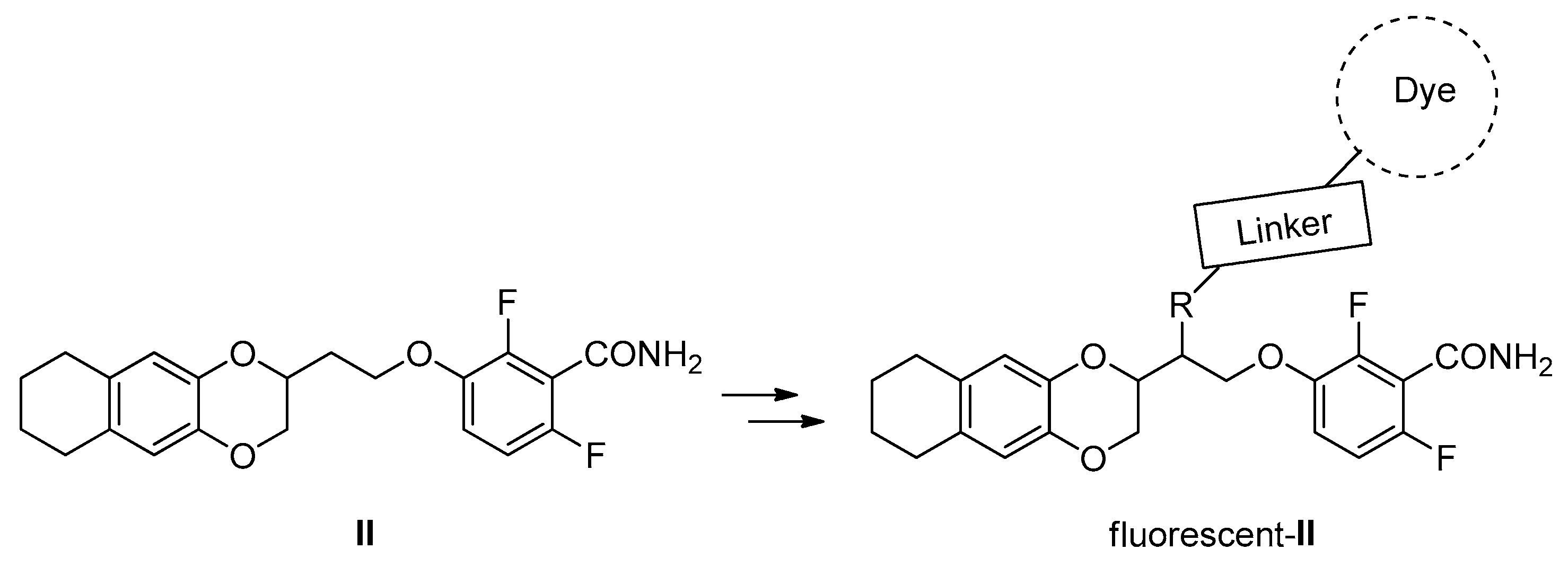
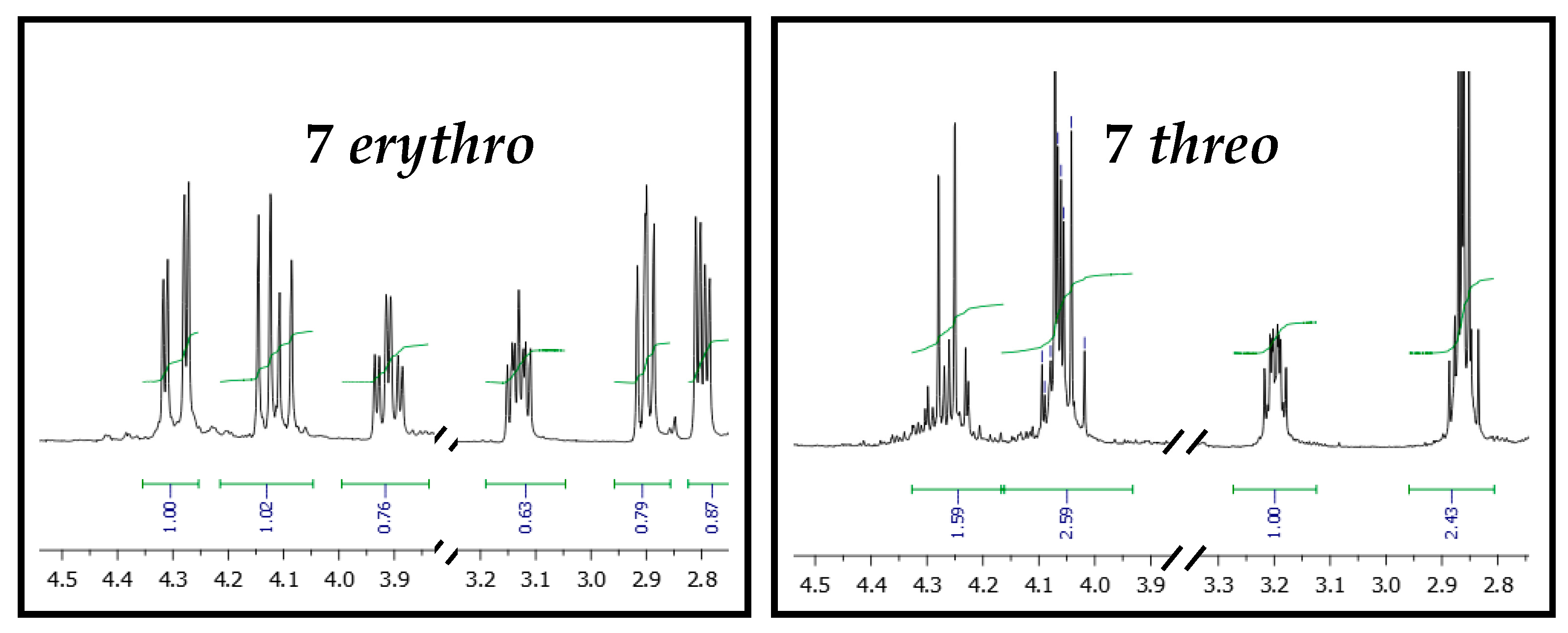
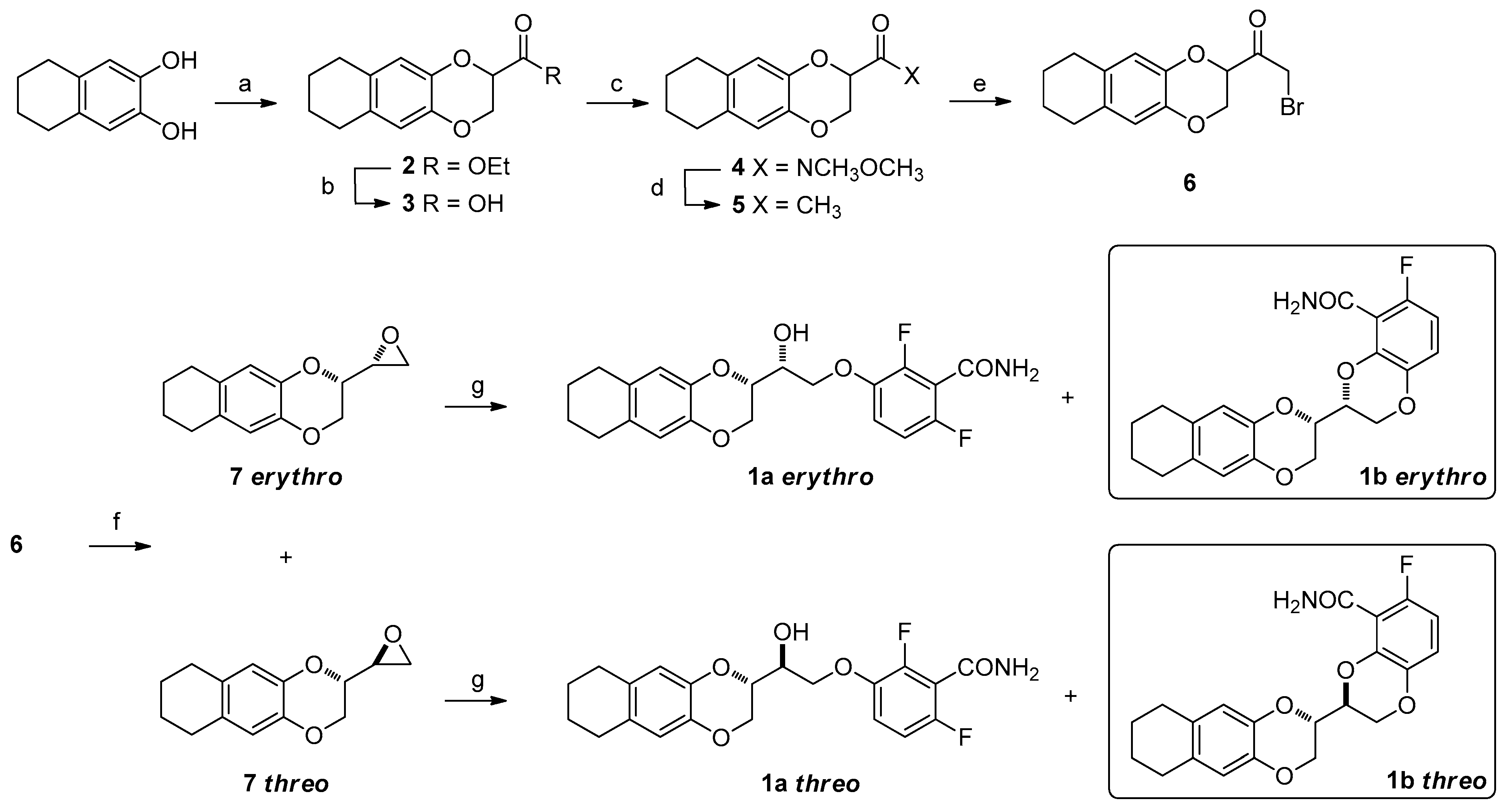
2. Results and Discussion
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- World Health Organization. Global Action Plan on Antimicrobial Resistance. 2015. Available online: https://www.who.int/publications/i/item/9789241509763 (accessed on 15 October 2022).
- World Health Organization. Antibiotic Resistance: Prevention and Control. Available online: https://www.who.int/news-room/fact-sheets/detail/antibiotic-resistance#:~:text=Antibiotic%20resistance%20is%20accelerated%20by,poor%20infection%20prevention%20and%20control (accessed on 15 October 2022).
- Pradhan, P.; Margolin, W.; Beuria, T.K. Targeting the Achilles Heel of FtsZ: The Interdomain Cleft. Front. Microbiol. 2021, 12, 732796. [Google Scholar] [CrossRef] [PubMed]
- Casiraghi, A.; Suigo, L.; Valoti, E.; Straniero, V. Targeting Bacterial Cell Division: A Binding Site-Centered Approach to the Most Promising Inhibitors of the Essential Protein FtsZ. Antibiotics 2020, 9, 69. [Google Scholar] [CrossRef] [PubMed]
- Straniero, V.; Pallavicini, M.; Chiodini, G.; Zanotto, C.; Volontè, L.; Radaelli, A.; Bolchi, C.; Fumagalli, L.; Sanguinetti, M.; Menchinelli, G.; et al. 3-(Benzodioxan-2-ylmethoxy)-2,6-difluorobenzamides bearing hydrophobic substituents at the 7-position of the benzodioxane nucleus potently inhibit methicillin-resistant Sa and Mtb cell division. Eur. J. Med. Chem. 2016, 120, 227–243. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.W.; Wu, L.J.; Czaplewski, L.G.; Errington, J. Multiple effects of benzamide antibiotics on FtsZ function. Mol. Microbiol. 2011, 80, 68–84. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.W.; Wu, L.J.; Errington, J. A benzamide-dependent ftsZ mutant reveals residues crucial for Z-ring assembly. Mol. Microbiol. 2016, 99, 1028–1042. [Google Scholar] [CrossRef]
- Haeusser, D.P.; Margolin, W. Splitsville: Structural and functional insights into the dynamic bacterial Z ring. Nat. Rev. Microbiol. 2016, 14, 305–319. [Google Scholar] [CrossRef]
- Carro, L. Recent Progress in the Development of Small-Molecule FtsZ Inhibitors as Chemical Tools for the Development of Novel Antibiotics. Antibiotics 2019, 8, 217. [Google Scholar] [CrossRef]
- Tripathy, S.; Sahu, S.K. FtsZ inhibitors as a new genera of antibacterial agents. Bioorg. Chem. 2019, 91, 103169. [Google Scholar] [CrossRef]
- Han, H.; Wang, Z.; Li, T.; Da, T.; Mao, R.; Hao, Y.; Yang, N.; Wang, X.; Wang, J. Recent progress of bacterial FtsZ inhibitors with a focus on peptides. FEBS J. 2021, 288, 1091–1106. [Google Scholar] [CrossRef]
- Elsen, N.L.; Lu, J.; Parthasarathy, G.; Reid, J.C.; Sharma, S.; Soisson, S.M.; Lumb, K.J. Mechanism of action of the cell-division inhibitor PC190723: Modulation of FtsZ assembly cooperativity. J. Am. Chem. Soc. 2012, 134, 12342–12345. [Google Scholar] [CrossRef]
- Fang, Z.; Li, Y.; Zheng, Y.; Li, X.; Lu, Y.-J.; Yan, S.-C.; Wong, W.-L.; Chan, K.-F.; Wong, K.-Y.; Sun, N. Antibacterial activity and mechanism of action of a thiophenyl substituted pyrimidine derivative. RSC Adv. 2019, 9, 10739–10744. [Google Scholar] [CrossRef]
- Straniero, V.; Sebastián-Pérez, V.; Suigo, L.; Margolin, W.; Casiraghi, A.; Hrast, M.; Zanotto, C.; Zdovc, I.; Radaelli, A.; Valoti, E. Computational Design and Development of Benzodioxane-Benzamides as Potent Inhibitors of FtsZ by Exploring the Hydrophobic Subpocket. Antibiotics 2021, 10, 442. [Google Scholar] [CrossRef]
- Chiodini, G.; Pallavicini, M.; Zanotto, C.; Bissa, M.; Radaelli, A.; Straniero, V.; Bolchi, C.; Fumagalli, L.; Ruggeri, P.; De Giuli Morghen, C.; et al. Benzodioxane-benzamides as new bacterial cell division inhibitors. Eur. J. Med. Chem. 2015, 89, 252–265. [Google Scholar] [CrossRef] [PubMed]
- Straniero, V.; Zanotto, C.; Straniero, L.; Casiraghi, A.; Duga, S.; Radaelli, A.; De Giuli Morghen, C.; Valoti, E. 2,6-Difluorobenzamide Inhibitors of Bacterial Cell Division Protein FtsZ: Design, Synthesis, and Structure-Activity Relationships. ChemMedChem 2017, 12, 1303–1318. [Google Scholar] [CrossRef]
- Straniero, V.; Sebastián Pérez, V.; Hrast, M.; Zanotto, C.; Casiraghi, A.; Suigo, L.; Zdovc, I.; Radaelli, A.; De Giuli Morghen, C.; Valoti, E. Benzodioxane-benzamides as antibacterial agents: Computational and SAR studies to evaluate the influence of the 7-substitution in FtsZ interaction. ChemMedChem 2020, 2, 195–209. [Google Scholar] [CrossRef]
- Straniero, V.; Suigo, L.; Casiraghi, A.; Sebastián-Pérez, V.; Hrast, M.; Zanotto, C.; Zdovc, I.; De Giuli Morghen, C.; Radaelli, A.; Valoti, E. Benzamide Derivatives Targeting the Cell Division Protein FtsZ: Modifications of the Linker and the Benzodioxane Scaffold and Their Effects on Antimicrobial Activity. Antibiotics 2020, 9, 160. [Google Scholar] [CrossRef] [PubMed]
- Artola, M.; Ruíz-Avila, L.B.; Ramírez-Aportela, E.; Martínez, R.F.; Araujo-Bazán, L.; Vázquez-Villa, H.; Martín-Fontecha, M.; Oliva, M.A.; Martín-Galiano, A.J.; Chacón, P.; et al. The structural assembly switch of cell division protein FtsZ probed with fluorescent allosteric inhibitors. Chem. Sci. 2017, 8, 1525–1534. [Google Scholar] [CrossRef] [PubMed]
- Ferrer-González, E.; Fujita, J.; Yoshizawa, T.; Nelson, J.M.; Pilch, A.J.; Hillman, E.; Ozawa, M.; Kuroda, N.; Al-Tameemi, H.M.; Boyd, J.M.; et al. Structure-Guided Design of a Fluorescent Probe for the Visualization of FtsZ in Clinically Important Gram-Positive and Gram-Negative Bacterial Pathogens. Sci. Rep. 2019, 9, 20092. [Google Scholar] [CrossRef] [PubMed]
- Stokes, N.R.; Baker, N.; Bennett, J.M.; Berry, J.; Collins, I.; Czaplewski, L.G.; Logan, A.; Macdonald, R.; Macleod, L.; Peasley, H.; et al. An improved small-molecule inhibitor of FtsZ with superior in vitro potency, drug-like properties, and in vivo efficacy. Antimicrob. Agents Chemother. 2013, 57, 317–325. [Google Scholar] [CrossRef]
- Stokes, N.R.; Baker, N.; Bennett, J.M.; Chauhan, P.K.; Collins, I.; Davies, D.T.; Gavade, M.; Kumar, D.; Lancett, P.; Macdonald, R.; et al. Design, synthesis and structure-activity relationships of substituted oxazole-benzamide antibacterial inhibitors of FtsZ. Bioorg. Med. Chem. Lett. 2014, 24, 353–359. [Google Scholar] [CrossRef]
- Casiraghi, A.; Valoti, E.; Suigo, L.; Artasensi, A.; Sorvillo, E.; Straniero, V. How Reaction Conditions May Influence the Regioselectivity in the Synthesis of 2,3-Dihydro-1,4-benzoxathiine Derivatives. J. Org. Chem. 2018, 83, 13217–13227. [Google Scholar] [CrossRef] [PubMed]
- Suigo, L.; Lodigiani, G.; Straniero, V.; Valoti, E. (3-Methylene-2,3-dihydronaphtho[2,3-b][1,4]dioxin-2-yl)methanol. Molbank 2022, 2022, M1521. [Google Scholar] [CrossRef]
- Straniero, V.; Casiraghi, A.; Fumagalli, L.; Valoti, E. How do reaction conditions affect the enantiopure synthesis of 2-substituted-1,4-benzodioxane derivatives? Chirality 2018, 30, 943–950. [Google Scholar] [CrossRef] [PubMed]
- Clark, R.D.; Caroon, J.M.; Kluge, A.F.; Repke, D.B.; Roszkowski, A.P.; Strosberg, A.M.; Baker, S.; Bitter, S.M.; Okada, M.D. Synthesis and antihypertensive activity of 4′-substituted spiro4H-3,1-benzoxazine-4,4′-piperidin-2(1H)-ones. J. Med. Chem. 1983, 26, 657–661. [Google Scholar] [CrossRef] [PubMed]
- Clark, R.D.; Kurz, L.J. Synthesis of the Enantiomers of Erythro-2-oxiranyl-1,4-benzodioxan. Heterocycles 1985, 23, 2005. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Straniero, V.; Suigo, L.; Lodigiani, G.; Valoti, E. Obtainment of Threo and Erythro Isomers of the 6-Fluoro-3-(2,3,6,7,8,9-hexahydronaphtho[2,3-b][1,4]dioxin-2-yl)-2,3-dihydrobenzo[b][1,4]dioxine-5-carboxamide. Molbank 2023, 2023, M1559. https://doi.org/10.3390/M1559
Straniero V, Suigo L, Lodigiani G, Valoti E. Obtainment of Threo and Erythro Isomers of the 6-Fluoro-3-(2,3,6,7,8,9-hexahydronaphtho[2,3-b][1,4]dioxin-2-yl)-2,3-dihydrobenzo[b][1,4]dioxine-5-carboxamide. Molbank. 2023; 2023(1):M1559. https://doi.org/10.3390/M1559
Chicago/Turabian StyleStraniero, Valentina, Lorenzo Suigo, Giulia Lodigiani, and Ermanno Valoti. 2023. "Obtainment of Threo and Erythro Isomers of the 6-Fluoro-3-(2,3,6,7,8,9-hexahydronaphtho[2,3-b][1,4]dioxin-2-yl)-2,3-dihydrobenzo[b][1,4]dioxine-5-carboxamide" Molbank 2023, no. 1: M1559. https://doi.org/10.3390/M1559
APA StyleStraniero, V., Suigo, L., Lodigiani, G., & Valoti, E. (2023). Obtainment of Threo and Erythro Isomers of the 6-Fluoro-3-(2,3,6,7,8,9-hexahydronaphtho[2,3-b][1,4]dioxin-2-yl)-2,3-dihydrobenzo[b][1,4]dioxine-5-carboxamide. Molbank, 2023(1), M1559. https://doi.org/10.3390/M1559