
3-(1-Ethylamino-ethylidene)-1-methyl-pyrrolidine-2,4-dione



Abstract
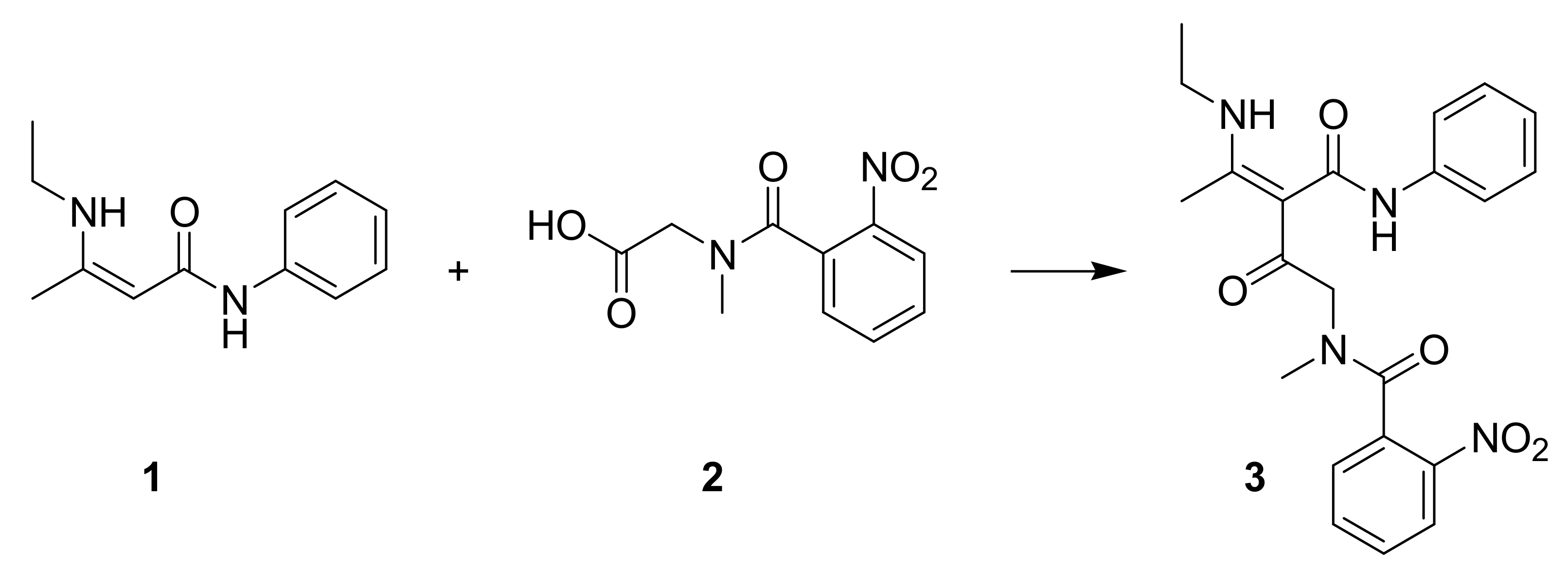
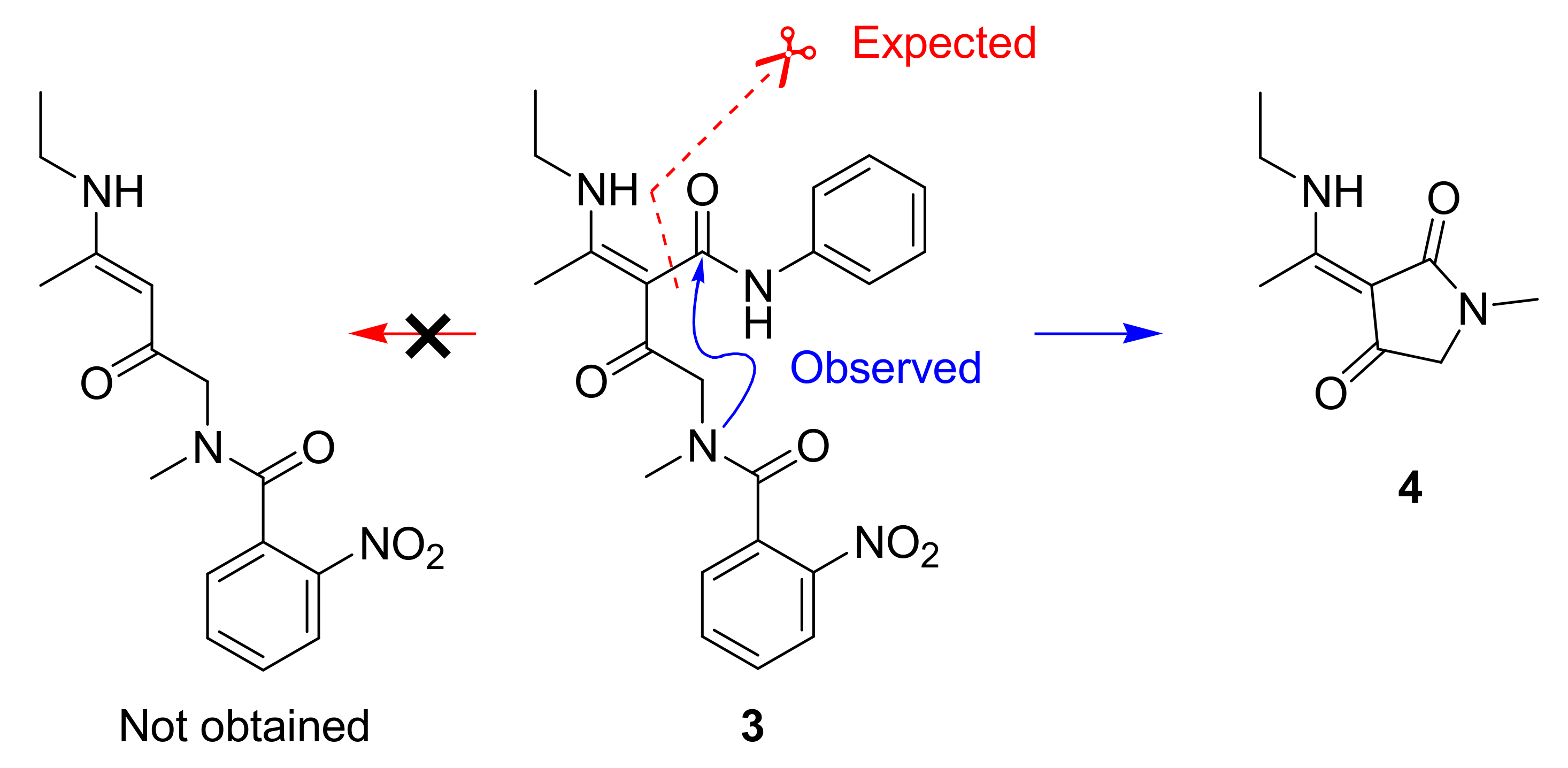
1. Introduction
2. Results
3. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mo, X.; Li, Q.; Ju, J. Naturally occurring tetramic acid products: Isolation, structure elucidation and biological activity. RSC Adv. 2014, 4, 50566–50593. [Google Scholar] [CrossRef]
- Petermichl, M.; Schobert, R. 3-Acyltetramic Acids: A Decades-Long Approach to a Fascinating Natural Product Family. Synlett 2017, 28, 654–663. [Google Scholar] [CrossRef]
- Schobert, R.; Schlenk, A. Tetramic and tetronic acids: An update on new derivatives and biological aspects. Bioorg. Med. Chem. 2008, 16, 4203–4221. [Google Scholar] [CrossRef] [PubMed]
- Jeong, Y.-C.; Moloney, M.G. Tetramic Acids as Scaffolds: Synthesis, Tautomeric and Antibacterial Behaviour. Synlett 2009, 2009, 2487–2491. [Google Scholar] [CrossRef]
- Burmeister, H.R.; Bennett, G.A.; Vesonder, R.F.; Hesseltine, C.W. Antibiotic Produced by Fusarium equiseti NRRL 5537. Antimicrob. Ag. Chemother. 1974, 5, 634–639. [Google Scholar] [CrossRef] [PubMed]
- Marfori, E.C.; Kajiyama, S.; Fukusaki, E.; Kobayashi, A. Trichosetin, a Novel Tetramic Acid Antibiotic Produced in Dual Culture of Trichoderma harzianum and Catharanthus roseus Callus. Z. Für Nat. C 2002, 57, 465–470. [Google Scholar] [CrossRef] [PubMed]
- Gänzle, M.G. Reutericyclin: Biological activity, mode of action, and potential applications. Appl. Microbiol. Biotechnol. 2004, 64, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, N.M.; Nazareth, J.; Divekar, P.V.; Kohl, H.; de Souza, N.J. Magnesidin, a novel magnesium-containing antibiotic. J. Antibiot. 1973, 26, 797–798. [Google Scholar] [CrossRef] [PubMed]
- Mathiadis, D.; Tsironis, D.; Stefanou, V.; Boussias, S.; Panagiotopoulou, A.; McKee, V.; Igglessi-Markopoulou, O.; Markopoulos, J. Synthesis, biological evaluation and structure-activity relationships of 5-arylidene tetramic acids with antibacterial activity against methicillin-resistant Staphylococcus aureus. Bioorg. Med. Chem. Lett. 2020, 30, 127107. [Google Scholar] [CrossRef] [PubMed]
- Linder, D.; Schobert, R. Synthesis of the fungus metabolite cladosin C. Org. Biomol. Chem. 2017, 15, 7672–7677. [Google Scholar] [CrossRef] [PubMed]
- Reber, K.P.; Mease, J.; Kim, J. Total Synthesis of Cladosins B and C. J. Org. Chem. 2020, 85, 11571–11578. [Google Scholar] [CrossRef] [PubMed]
- Yuki, H.; Kaizu, Y.; Yoshida, S.; Higuchi, S.; Honda, S.; Takiura, K. Studies of Tenuazonic Acid Analogs. I. Synthesis of 5-Substituted 3-(1’-Anilinoethylidene) pyrrolidine-2, 4-dione. Chem. Pharm. Bull. 1971, 19, 1664–1668. [Google Scholar] [CrossRef]
- Jeong, Y.-C.; Anwar, M.; Moloney, M.G. Synthesis, antibiotic activity and structure–activity relationship study of some 3-enaminetetramic acids. Bioorg. Med. Chem. Lett. 2014, 24, 1901–1906. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.-F.; Si, T.-F.; Li, Q.-B.; Zhu, X.-J.; Qiang, S.; Yang, C.-L. Synthesis, characterization and biological activity of novel (5-RS,6-S)-5-sec-butyl-3-(1-substituted-amino)-ethylidene-1H-pyrrolidine-2,4-diones. ARKIVOC 2010, 2010, 31–48. [Google Scholar] [CrossRef]
- Angelov, P.; Ivanova, S.; Yanev, P. Enamines of 3-acyltetramic acids from β-enamino amides and amino acids. Tetrahedron Lett. 2017, 58, 4776–4778. [Google Scholar] [CrossRef]
- Venkov, A.P.; Angelov, P.A. Synthesis of Unsymmetrical β-Enamino Ketones. Synthesis 2003, 2003, 2221–2225. [Google Scholar] [CrossRef]
- Tietze, O.; Schiefner, B.; Ziemer, B.; Zschunke, A. Assignment of E/Z isomers in Schiff bases of 3-acyltetramic acids with ethylenediamine by 1H and 13C NMR spectroscopy. Fresenius. J. Anal. Chem. 1997, 357, 477–481. [Google Scholar] [CrossRef]
- Gavrielatos, E.; Markopoulos, J.; Igglessi-Markopoulou, O. Synthesis and NMR spectroscopic studies of novel N-acetyl-3-aminoalkyl tetramic acids. Heterocyclic Comm. 1999, 5, 515–520. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| General Formula | R | Inhibition Zone (mm) | |
|---|---|---|---|
| E. coli ATCC 25922 | S. aureus ATCC 25923 | ||
 | CH3 | 17 | 15 |
| COOCH2CH3 | - | - | |
| COOCH2CCl3 | - | - | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Angelov, P.; Terziyska, P.; Georgiev, D. 3-(1-Ethylamino-ethylidene)-1-methyl-pyrrolidine-2,4-dione. Molbank 2023, 2023, M1556. https://doi.org/10.3390/M1556
Angelov P, Terziyska P, Georgiev D. 3-(1-Ethylamino-ethylidene)-1-methyl-pyrrolidine-2,4-dione. Molbank. 2023; 2023(1):M1556. https://doi.org/10.3390/M1556
Chicago/Turabian StyleAngelov, Plamen, Paraskeva Terziyska, and Danail Georgiev. 2023. "3-(1-Ethylamino-ethylidene)-1-methyl-pyrrolidine-2,4-dione" Molbank 2023, no. 1: M1556. https://doi.org/10.3390/M1556
APA StyleAngelov, P., Terziyska, P., & Georgiev, D. (2023). 3-(1-Ethylamino-ethylidene)-1-methyl-pyrrolidine-2,4-dione. Molbank, 2023(1), M1556. https://doi.org/10.3390/M1556